Etiology and Pathophysiology
In FMF, it is thought that a mutated pyrin is associated with an abnormal activation of the interleukin-1 (IL-1) pathway, resulting in uncontrolled inflammation. This disease can have profound effects on the innate immune pathway.
More than 50 mutations in the Mediterranean fever (MEFV) gene have been associated with FMF.2 The MEFV gene consists of 10 exons, and most of the disease-causing mutations are located in exon 10. The gene codes for pyrin, an important protein of the inflammasome and the inflammatory pathway. Pyrin regulates IL-1 production through its interaction with caspase-1; Chae et al have suggested a direct, ASC (apoptosis-associated speck-like protein with a caspase-recruitment domain)-independent effect of pyrin on IL-1β activation and a direct interaction of pyrin with caspase-1 to modulate IL-1β production.7 Thus, mutations in pyrin are associated with increased IL-1β production. Importantly, the IL-1 receptor antagonist anakinra can suppress acute-phase proteins in a patient with FMF and amyloidosis.7
Clinical Features
FMF typically manifests in childhood with recurrent, self-limited attacks of fever in association with abdominal pain, arthritis, or chest pain; these clinical manifestations can occur alone or together.1-3,8,9 The clinical hallmark is an attack in the form of fever and serositis along with elevation in acute phase reactants. The typical attack of FMF, which lasts one-half to three days, is accompanied by fever.1,3,8 Myalgia may occur.9 In the majority of the patients, serositis manifests itself as abdominal pain which can be incapacitating. Arthritis or arthralgia is also very frequent and was present in half of the patients in a large series reported from Israel and Turkey.8,9 Pleurisy in the form of chest pain is common; it is usually unilateral and often spreads to the shoulder.
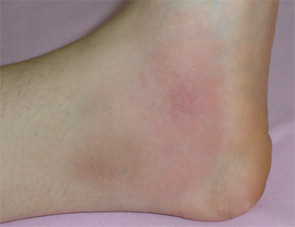
Rash is not expected in FMF except when it is associated with vasculitis and the erysipelas-like erythema (ELE) which is provoked by pressure-bearing exercise (see Figure 1, p. 20). ELE may not necessarily accompany an attack. In fact, some manifestations of FMF are not associated with attacks. These include protracted febrile myalgia, hematuria, arthralgia, and exertional leg pain.8 Another notable feature of FMF is the association with vasculitis and, especially, polyarteritis nodosa.10 A large series from Turkey has shown that FMF patients have increased rates of Henoch-Schonlein purpura, polyarteritis nodosa, and probably Behçet disease.9
Laboratory Findings
While spontaneous rises and falls of erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), and fibrinogen are quite characteristic of the attacks in FMF, substantial subclinical inflammation occurs widely and over prolonged periods in patients with FMF.10,11 These findings suggest that the relatively infrequent clinically overt attacks represent the tip of the iceberg in this disorder: Lachman et al have shown that there was also inflammatory activity between attacks as well (median serum amyloid A [SAA] 6mg/l, CRP 4 mg/l).11 SAA levels have proven to be the most informative in identifying the level of inflammation. In a study from our center, my colleagues and I have shown that SAA levels are also associated with an insufficient dose of colchicine, and we use SAA levels to monitor disease control.
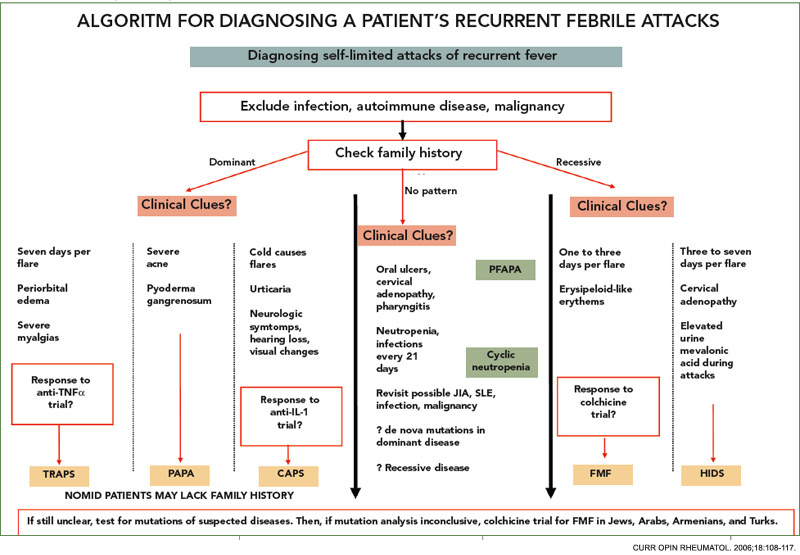
FMF is clinically diagnosed on the basis of typical attacks with an increased inflammatory response. Livneh et al from Israel, have suggested a set of criteria for diagnosis; however, these have not been validated in other ethnic groups.12 According to these guidelines, there are major and minor criteria as well as supportive criteria. The four major criteria include the typical attacks (≥3 of the same type; rectal temperature ≥38°C; lasting 12 to 72 hours) with peritonitis, pleuritis, monoarthritis (hip, knee, ankle), and fever alone. Minor criteria are defined as incomplete attacks, exertional leg pain, and favorable response to colchcine.12 The definite confirmation or the “gold standard” for diagnosis is the identification of two disease-causing mutations.
When the gene for FMF was identified in 1997, we thought that our problems with diagnosis were over and that we would rely on genetics for diagnosis. However, the experience in Israel and Turkey has shown that we are not able to identify two mutations (which is required to confirm the diagnosis in an autosomal recessive disease), in one-quarter to one-third of the patients. This may be due to the existence of mutations that are not routinely screened or the possibility of mutations in the promoter region of the pyrin gene. However, the explanation may be more complicated due to the contribution of other genetic factors. One also needs to consider the possibility of a wrong diagnosis with the presence of other periodic fever syndromes, discussed below. We were left with the question of what to do with the following type of patient, who has a finding of FMF but does not have genetic evidence of this disease.
Case 2: An 11-year-old boy was referred to us by the local physician with a probable diagnosis of FMF. The boy had had two attacks of fever and severe incapacitating abdominal pain and a recent attack of monoarthritis in the ankle. He occasionally complained of pain in his calf muscles with exercise. A cousin was on dialysis for secondary renal amyloidosis. The boy had no features suggestive of the other autoinflammatory syndromes. We tested him and found him to carry a M680I mutation in a single allele and no mutations in the other. Should we tell him to take colchicine thrice daily for the rest of his life?
This patient has a very strong history for FMF and there is a case of secondary amyloidosis in the family. Our approach would be to test his acute phase reactants and SAA levels and put him on a trial of colchicine. FMF is the only autoinflammatory disease that responds to colchicine. Thus, the trial period with colchicine would be followed by a period of withdrawal of the drug where we observe the symptoms and the SAA levels. We would then decide whether to accept this patient to have familial Mediterranean fever based on this data.
The differential diagnosis of FMF is quite large (see Table 1, above). Ethnicity is an important factor, although it must be remembered that FMF has been encountered in all groups, including Japanese. All autoinflammatory syndromes have a number of characteristic features which should be remembered in dealing with a patient with a periodic fever syndrome.
Differential Diagnosis
CIAS-1-pathies cryopyrin-associated periodic syndromes (CAPS): Three diseases have been associated with mutations in this group: the familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), and neonatal-onset multi-system inflammatory disease (NOMID, also known as chronic infantile neurologic cutaneous articular [CINCA] syndrome). CIAS-1-pathies, or CAPS, are a group of autoinflammatory diseases caused by mutations in the gene coding for cryopyrin.2,5,13,14 Cryopyrin has a pyrin domain and is a part of the macromolecular complex called the inflammasome which mediates IL-1β activation.
Among these three major diseases, FCAS represents the mildest and NOMID the most severe form of the disease complex.14 All are characterized by fevers and a non-pruritic, urticarial rash, usually presenting in infancy (or later in MWS), and they may have some overlapping features, described below. FCAS presents with attacks precipitated by cold.13 The main differentiating feature of MWS is a hearing defect. The names of the third disease in this group reflects its characteristics; NOMID for the onset and CINCA for the involved systems. NOMID, and sometimes the other diseases, tend to have more persistent features and may lack the typical attacks which may prove important in differential diagnosis.13,14
Hyperimmunoglobulenemia-D with periodic fever syndrome (HIDS): HIDS is an autosomal, recessively inherited, periodic fever syndrome caused by mutations in the gene MVK, which codes for the enzyme mevalonate kinase.15,16 The majority of reported patients are from The Netherlands and France. However, cases have been reported worldwide. The attacks typically last for two to seven days. Along with the fever, abdominal pain, rashes, diarrhea, vomiting, and cervical lymphadenopathy are common.15,16 Arthralgia and arthritis and hepatosplenomagaly may occur.
Tumor necrosis factor receptor–associated periodic syndrome (TRAPS): This autosomal dominantly inherited disease is associated with mutations on chromosome 12 in TNFRSF1A, the gene encoding the 55-kDa receptor for TNF.17 The attacks last longer than a week, and muscle involvement is a frequent feature in these patients, usually affecting only one muscle group at a time.
Other rare diseases: Pyogenic sterile arthritis, pyoderma gangrenosum, and acne syndrome (PAPA), Crohn disease, and Blau’s syndrome are among the other monogenic autoinflammatory diseases. Caspase associated recruitment domains (CARD) 15 mutations have been shown in 40% of Crohn’s, 50% of Blau’s, and 90% of early-onset sarcoidosis patients.2 CARD is also a part of the inflammasome.
Autoinflammatory syndromes that are not monogenic and autoinflammatory diseases with complex genetic traits: This spectrum has been growing and is claimed to include diseases such as Behçet Disease, systemic juvenile idiopathic arthritis.18
Management Principles and Prognosis
The use of colchicine has been a breakthrough in the management of FMF. This is the only periodic fever where an excellent quality of life is possible in the majority of patients—and with a cheap pill. In fact, colchicine has been shown to prevent amyloidosis in compliant patients in series from Israel and Turkey.19,20 In a large pediatric study (n=809), no manifestations of amyloidosis occurred in 809 treated children during an observation period of up to 13 years.19 In the Turkish FMF registry, 2.3% developed amyloidosis, all being noncompliant with respect to regular colchicine intake.20 We do not know the genetic factors that determine the development of secondary amyloidosis, although a number of polymorphisms in relevant genes have been associated with the development of secondary amyloidosis in FMF.
A starting dose of 0.5 mg/day colchicine in young children and 1.0 mg/day in older children (older than five years) is sufficient for disease control in approximately half of patients.21 This is increased to 1.5–2 mg/day once the child is more than a square meter. SAA levels might indicate insufficient disease control and thereby require dose adjustment.22
As physicians, we should warn the families about the risk to other children and follow patients’ siblings closely for any symptoms of unexplained fever or typical attacks. A routine mutation analysis of the FMF gene for siblings is probably not indicated. However, if there is a case of renal amyloidosis in the family, a genetic workup may be informative. SAA levels may need to be checked in patients with one mutation only and in siblings.
Dr. Ozen is professor of pediatrics at Hacettepe University in Ankara, Turkey.
References
- Livneh A, Langevitz P. Diagnostic and treatment concerns in familial Mediterranean fever. Baillieres Best Pract Res Clin Rheumatol. 2000;14(3):477-498.
- Stojanov S, Kastner DL. Familial autoinflammatory diseases: genetics, pathogenesis and treatment. Curr Opin Rheumatol. 2005;17(5):586-599.
- Ozen S. Familial mediterranean fever: revisiting an ancient disease. Eur J Pediatr. 2003;162(7-8):449-454.
- Aksentijevich I, Torosyan Y, Samuels J, et al. Mutation and haplotype studies of familial Mediterranean fever reveal new ancestral relationships and evidence for a high carrier frequency with reduced penetrance in the Askhenazi Jewish population. Am J Hum Genet. 1999; 64: 949-962.
- Samuels J, Ozen S. Familial Mediterranean fever and the other autoinflammatory syndromes: Evaluation of the patient with recurrent fever. Curr Opin Rheumatol.2006;18:108-117.
- Yilmaz E, Ozen S, Balci B, et al. Mutation frequency of Familial Mediterranean Fever and evidence for a high carrier rate in the Turkish population. Eur J Hum Genet. 2001; Jul; 9(7):553-555.
- Chae JJ, Wood G, Masters SL, Richard K, et al. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci U S A. 2006 Jun 27;103(26):9982-9987.
- Livneh A, Langevitz P, Zemer D, et al. The changing face of familial Mediterranean fever. Semin Arthritis Rheum. 1996;26(3): 612-627.
- Tunca M, Akar S, Onen F, et al. Familial Mediterranean fever (FMF) in Turkey: results of a nationwide multicenter study. Medicine (Baltimore). 2005;84(1):1-11
- Ozen S, Bakkaloglu A, Yilmaz E, et al. Mutations in the gene for familial Mediterranean fever: do they predispose to inflammation? J Rheumatol. 2003 Sep;30(9):2014-2018.
- Lachmann HJ, Sengul B, Yavuzsen TU, et al. Clinical and subclinical inflammation in patients with familial Mediterranean fever and in heterozygous carriers of MEFV mutations. Rheumatology. 2006; 45(6):746-750
- Livneh A, Langevitz P, Zemer D, et al. (1997) Criteria for the diagnosis of FMF. Arthritis Rheum.1997;40:1879-1885.
- Hoffman HM, Rosengren S, Boyle DL. et al. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet. 2004;364(9447): 1779-1785.
- Neven B, Callebaut I, Prieur AM, et al. Molecular basis of the spectral expression of CIAS1 mutations associated with phagocytic cell-mediated autoinflammatory disorders CINCA/NOMID, MWS, and FCU. Blood. 2004;103:2809-2815
- Drenth JP, Cuisset L, Grateau G, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet. 1999;22(2):178-181.
- Frenkel J, Houten SM, Waterham HR, et al. Clinical and molecular variability in childhood periodic fever with hyperimmunoglobulinaemia D. Rheumatology (Oxford). 2001 May; 40(5):579-584.
- McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell.1999; 97(1):133-144.
- Ozen S, Hoffman HM, Frenkel J, Kastner D. Familial Mediterranean fever (FMF) and beyond: a new horizon. Fourth International Congress on the Systemic Autoinflammatory Diseases held in Bethesda, USA, 6-10 November 2005. Ann Rheum Dis. 2006; 65(7):961-964.
- Zemer D, Livneh A, Danon YL, Pras M, Sohar E. Long-term colchicine treatment in children with familial Mediterranean fever. Arthritis Rheum. 1991 Aug;34(8): 973-977.
- Saatci U, Ozen S, Ozdemir S, et al. Familial Mediterranean fever in children: report of a large series and discussion of the risk and prognostic factors of amyloidosis. Eur J Pediatr. 1997; 156(8):619-623.
- Kallinich T, Haffner D, Niehues T, et al. Colchicine use in children and adolescents with familial Mediterranean fever: literature review and consensus statement. Pediatrics. 2007;119(2):474-483.
- Berkun Y, Padeh S, Reichman B, et al. A single testing of serum amyloid a levels as a tool for diagnosis and treatment dilemmas in familial Mediterranean fever. Semin Arthritis Rheum. 2007;37:182-188.