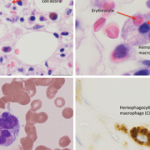
In pediatric rheumatology, the term macrophage activation syndrome (MAS) refers to a condition caused by excessive activation and expansion of T lymphocytes and macrophagic histiocytes that exhibit hemophagocytic activity.1-4 Although the pathognomonic feature of MAS (i.e., histiocytes phagocytosing normal hematopoietic elements) is usually seen in bone marrow (see Figure 1, p. 23), such cells can infiltrate almost any organ in the body. The expansion of these cells also leads to a massive systemic inflammatory response associated with three cardinal features: cytopenias, liver dysfunction, and coagulopathy resembling disseminated intravascular coagulation (DIC). MAS is a life-threatening condition, and the reported mortality rates reach 20–30%.
Although MAS has been reported in association with almost any rheumatic disease, it is by far most common in systemic juvenile idiopathic arthritis (SJIA). Conversely, about 10% of patients with SJIA develop full-blown MAS, while mild “subclinical” MAS may be seen in as many as one-third of patients with active systemic disease.3,5 Besides SJIA, systemic lupus erythematosus (SLE), and Kawasaki disease are two other rheumatologic conditions in which MAS appears to occur somewhat more frequently than in other diseases. Although most patients develop this syndrome sometime during the course of their primary rheumatic disease following diagnosis, MAS occurring at the initial presentation of a rheumatic illness is rather common. In adults, based on some limited epidemiologic studies, MAS is seen most frequently in association with adult onset Still’s disease, SLE, and various vasculitic syndromes.
Because expansion of tissue macrophages, or histiocytes, exhibiting hemophagocytic activity in MAS is often triggered by infections or modifications in the drug therapy, the term reactive hemophagocytic lymphohistiocytosis has been preferred by some authors to classify this condition.6 This term suggests that MAS may belong to a group of histiocytic disorders collectively known as hemophagocytic lymphohistiocytosis (HLH). HLH is a more general term that describes a spectrum of diseases characterized by accumulations of histologically benign well-differentiated mononuclear cells with a macrophage phenotype.7-9 Because such macrophages represent a subset of histiocytes that are distinct from Langerhans cells, this entity should be distinguished from Langerhans cell histiocytosis as well as other dendritic cell disorders. In the current classification of histiocytic disorders, HLH is further subdivided into primary or familial HLH and secondary or reactive HLH. Clinically, however, these conditions may be difficult to distinguish from each other. Familial hemophagocytic lymphohistiocytosis (FHLH) is a constellation of rare autosomal recessive immune disorders. The clinical symptoms of FHLH usually become evident within the first two months of life. The course of disease is very severe, and most patients eventually require hematopoietic stem cell transplantation. Secondary HLH tends to occur in older children and more often is associated with an identifiable infectious episode, most notably Epstein-Barr virus (EBV) or cytomegalovirus (CMV) infection. The group of secondary hemophagocytic disorders also includes malignancy associated HLH. Although there are many clinical similarities between MAS and both primary and secondary HLH, the exact relationship between these conditions is not understood.
TABLE 1: HLH-2004: Revised diagnostic guidelines for HLH10
The diagnosis HLH can be established if one of the two criteria below is met:
- A molecular diagnosis consistent with HLH (i.e., reported mutations found in either PRF1 or MUNC13-4); or
- Diagnostic criteria for HLH are fulfilled (i.e., at least five of the eight criteria listed below are present:
- Persistent fever
- Splenomegaly
- Cytopenias (affecting ≥2 of 3 lineages in the peripheral blood):
- Hemoglobin <90g/L (in infants <4 weeks: <100g/L)
- Platelets <100 x 109/L
- Neutrophils <1.0 x 109/L
- Hypertriglyceremia and/or hypofibrinogenemia:
- Fasting triglycerides ≥3.0 mmol/L (i.e., ≥ 265mg/dl)
- Fibrinogen ≤1.5 g/L
- Hemophagocytosis in bone marrow* or spleen or lymph nodes, no evidence of malignancy
- Serum ferritin ≥ 500µg/L (i.e., 500 ng/ml)
- Low or absent NK cell activity (according to local laboratory reference)
- Increased serum sIL2Rα (according to local laboratory reference)
* If hemophagocytic activity is not proven at the time of presentation, further search for hemophagocytic activity is encouraged. If the bone marrow specimen is not conclusive, material may be obtained from other organs.