 In September 2019, nintedanib became the first treatment approved by the U.S. Food & Drug Administration (FDA) to slow the rate of decline in pulmonary function of patients with systemic sclerosis-associated interstitial lung disease (SSc-ILD).1 FDA approval of tocilizumab for the same indication followed in March 2021.2
In September 2019, nintedanib became the first treatment approved by the U.S. Food & Drug Administration (FDA) to slow the rate of decline in pulmonary function of patients with systemic sclerosis-associated interstitial lung disease (SSc-ILD).1 FDA approval of tocilizumab for the same indication followed in March 2021.2
As rheumatology providers, we can all agree that additional treatment options for systemic sclerosis (SSc)—especially those with FDA approval—are a welcome change to the SSc treatment landscape. However, no one-size-fits-all approach exists for the care of patients with SSc and pulmonary involvement. SSc and SSc-ILD are clinically heterogeneous, and optimal pulmonary monitoring and therapeutic strategies are not yet clearly defined.

Dr. Hummers
In January 2022, Khanna et al. published a clinically relevant review on the diagnosis and treatment of SSc-ILD and proposed a clinical approach for risk stratification and therapeutic management in a clinical context. In this article, we discuss their recommendations and offer additional input from another SSc expert, Laura K. Hummers, MD, co-director, Johns Hopkins Scleroderma Center, associate professor of medicine, Division of Rheumatology, Johns Hopkins University, Baltimore.3
Background
SSc is a heterogenous autoimmune disease with the highest individual mortality of all rheumatic conditions.4 Over the past decade, research has focused on defining clinical subtypes of SSc to predict disease course and better tailor therapies. Clinical subtypes are stratified by degree of skin involvement (i.e., limited vs. diffuse) and autoantibody seropositivity.5
Given the efficacy of angiotensin-converting enzyme inhibitors in scleroderma renal crisis, SSc-ILD is now among the leading causes of SSc-related death.6 The prevalence of SSc-ILD depends on multiple factors, such as screening strategy (i.e., high-resolution computed tomography [HRCT] vs. pulmonary function tests [PFTs]); however, national observational registries and international cohorts show that approximately 65% of SSc patients have or will develop SSc-ILD at some point during their disease course.7,8
In 2006 and 2016, the results of two scleroderma lung studies changed practice patterns in SSc-ILD after demonstrating the benefits of cyclophosphamide (CYC) and mycophenolate mofetil (MMF). However, neither treatment has been FDA approved for this indication to date.9,10
Regarding FDA-approved therapies, the Safety and Efficacy of Nintedanib in SSc (SENSCIS) trial showed the agent, an anti-fibrotic tyrosine kinase inhibitor, slowed the rate of decline in pulmonary function in patients with SSc-ILD.11 Tocilizumab, an anti-interleukin 6 receptor antagonist, was studied in patients with SSc and early, diffuse cutaneous disease and elevated acute phase reactants. Although the primary end point for improvement in skin fibrosis was not met, forced vital capacity (FVC) data indicated that tocilizumab may preserve lung function in this population.12 A follow-up trial demonstrated that preservation of percent predicted FVC was greater with tocilizumab treatment than placebo, leading to the FDA approval.13
Diagnosis & Screening of SSc-ILD
Khanna et al. reviewed the evidence supporting the use of both HRCT and complete PFT for the initial screening and diagnosis of SSc-ILD.14 They recommend baseline HRCT and PFT in all patients with SSc. They reasoned that PFTs alone aren’t adequate because values may be normal early in the course of disease.6
Dr. Hummers offers a slightly different take. “At our center, all SSc patients get baseline and follow-up PFTs to evaluate for evolving SSc-ILD and pulmonary hypertension,” she says. “We don’t perform HRCT on all patients because there are lower risk subsets for whom this likely isn’t necessary. However, this is a nuanced decision, and it may be safer to just say everyone needs one.
“My real concern is that routine HRCT may lead to low-risk patients with mild SSc-ILD receiving unnecessary, expensive and potentially toxic treatment. Let’s say you refer a patient with limited cutaneous disease, anti-centromere antibody positivity and mild SSc-ILD to a pulmonologist. This patient is probably at very low risk of ILD progression, but pulmonologists may think about these patients as similar to the patients with idiopathic pulmonary fibrosis they treat most often. Nintedanib could be prescribed … [for a] patient [who should never be exposed to [it].”
Differentiation & Risk Stratification of SSc-ILD
Khanna et al. recommend the differentiation of subclinical from clinical SSc-ILD and risk stratification of patients at low vs. high risk of SSc-ILD progression (see Figure 1, below). These terms are defined as:
- Subclinical SSc-ILD: an absence of clinical symptoms, minimal SSc-ILD findings on HRCT and a normal or stable FVC;
- Clinical SSc-ILD: clinical symptoms with either SSc-ILD on HRCT and/or abnormal or clinically meaningful decline in FVC or diffusing capacity of the lungs for carbon monoxide (DLCO); and
- High risk of progressive SSc-ILD: patients with progressive skin disease, anti-topoisomerase I (anti-Scl-70) antibody positivity or elevated acute phase reactants.
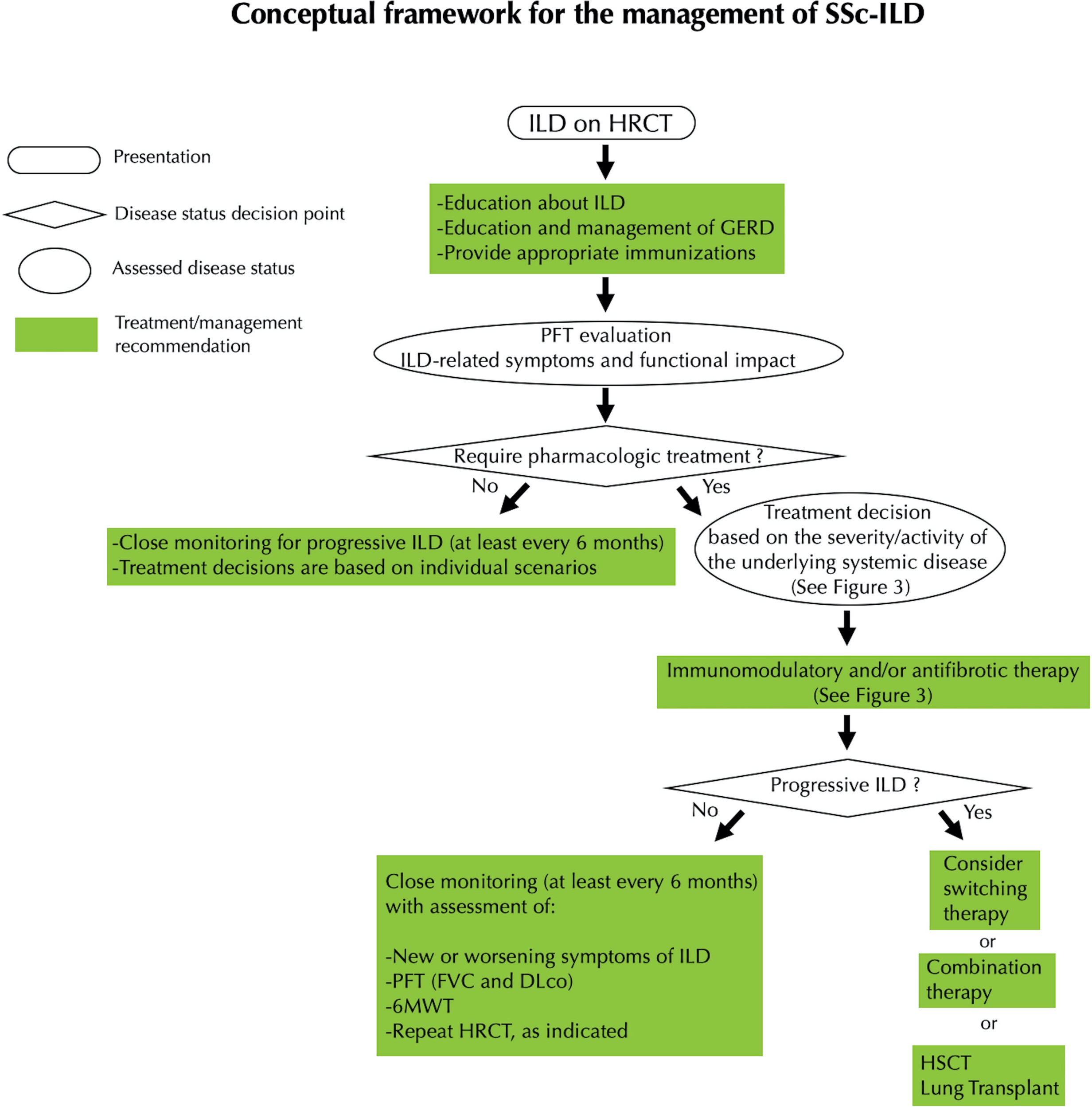
FIGURE 1 (click to enlarge): Expert Opinion on the Management of SSc-ILD
Reprinted from Khanna et al. by permission of Wiley.
Legend: Conceptual framework for the management of systemic sclerosis–associated interstitial lung disease (SSc‐ILD). HRCT = high‐resolution computed tomography; GERD = gastroesophageal reflux disease; PFT = pulmonary function test; FVC = forced vital capacity; DLco = diffusing capacity for carbon monoxide; 6MWT = 6‐minute walk test; HSCT = hematopoietic stem cell transplantation.
Khanna et al. recommend treatment for patients with clinical SSc-ILD or subclinical SSc-ILD with a high risk of disease progression. Those with subclinical SSc-ILD with low risk of progression require close monitoring (at least every six months) to confirm stability. Monitoring should involve assessment for new or worsening symptoms, PFTs (FVC and DLCO), a six-minute walk test and HRCT as indicated.
Initial Treatment of SSc-ILD
If treatment is indicated for a patient, which therapy should we choose? It’s truly the dawn of a new era in rheumatology when the question is not “What drug?” but which to use. Khanna et al. recommend basing treatment decisions on the presence of extrapulmonary manifestations of disease. Dr. Hummers agrees with this approach, noting “the treatment approach for clinical SSc-ILD is similar at our institution.”
For clinical SSc-ILD without extrapulmonary manifestations, the authors recommend MMF as a first-line treatment given its favorable toxicity profile compared with CYC. They also list nintedanib as an option.
Dr. Hummers practices similarly, using MMF as a first-line treatment. If patients are unable to tolerate MMF, she’ll switch them to nintedanib if there are no extrapulmonary manifestations of disease that warrant use of a different immunomodulatory drug.
For clinical SSc-ILD without extrapulmonary manifestations, Khanna et al. recommend mycophenolate mofetil as a first-line treatment given its favorable toxicity profile compared with cyclophosphamide.
“Of note, if patients don’t tolerate MMF due to GI [gastrointestinal] side effects, the chances of them tolerating nintedanib are low, in my experience. Patients often run into the same GI side effects with nintedanib,” she says.
For clinical SSc-ILD with active extrapulmonary manifestations, the authors recommend MMF or tocilizumab as a first-line treatment. CYC and rituximab are also options. They note that up-front combination therapy with MMF and nintedanib may be considered in patients with rapidly progressive pulmonary disease. Nintedanib monotherapy is not recommended given a lack of proven benefit for skin or musculoskeletal manifestations of disease.
“I select a therapy based on what will treat the most symptoms,” says Dr. Hummers. “If they have lung plus skin or lung plus muscle [involvement], I opt for MMF. If they have lung plus joints, I’d consider tocilizumab. However, the problem with tocilizumab is that the population studied was so narrow. These were patients very early in their disease course with diffuse cutaneous disease. They didn’t all have ILD, and you had to have elevated acute phase reactants to be included in the trial. So it’s tough to know what the true impact of tocilizumab is on lung disease outside of this narrow target population.”
For subclinical SSc-ILD at high risk of progressive disease, the authors recommend tocilizumab as a first-line therapy given the evidence from tocilizumab trials.14 MMF and CYC are also listed as options; however, randomized controlled data do not currently exist to support the use of one treatment over the other in this regard.
This last bit is where Dr. Hummers’ approach differs the most from that suggested by Khanna et al. “I would argue that there are still some people in this group [subclinical SSc-ILD at high risk of progressive disease] who I would still just watch,” she says. “For example, patients with limited cutaneous disease, anti-centromere antibody positive, elevated acute phase reactants and mild ILD; or patients with limited cutaneous disease, anti-Scl-70 positivity and mild ILD. I wouldn’t automatically reach for tocilizumab [for] these patients, especially given the narrow population studied in the trials, their expense and the potential for toxicity.”
Treatment of Progressive SSc-ILD
Should a patient’s pulmonary disease progress while they are on the initial regimen, the authors recommend:
- Switching therapies;
- Considering combination immunomodulatory therapy (e.g., MMF plus tocilizumab) or adding nintedanib; and
- Considering hematopoietic stem cell transplant or lung transplant.
Dr. Hummers agrees. When it comes to adding nintedanib, she says, “I am almost always using it in someone who has or is at risk for progressive ILD. In the nintedanib trial, a subset analysis showed the lowest amount of lung function decline was in those taking combination MMF and nintedanib. The study wasn’t powered to look at the effect of combination therapy, but we can infer that combo therapy may be beneficial.”
Future Directions
Regarding the future direction of research in SSc-ILD, Dr. Hummers says, “For me, there are two burning questions: 1) Outside the narrow population studied in the tocilizumab trials, is there any role for tocilizumab in ILD?; and 2) Is there a population of patients who should get combination MMF and nintedanib up front?”
In summary, Khanna et al. provide a practical approach to the care of patients with SSc-ILD that serves as a valuable resource to practicing rheumatology providers.
Dr. Hummers offers additional expert insight on the impact and incorporation of new trial data on clinical practice.
Treatment choices should consider disease severity, risk of progression and extrapulmonary disease activity. Baseline PFTs should be acquired in all patients with SSc, with consideration of HRCT in most patients. In those with subclinical SSc-ILD at high risk of progression, treatment may also be considered.
Samantha C. Shapiro, MD, is an academic rheumatologist and an affiliate faculty member of the Dell Medical School at the University of Texas at Austin. She received her training in internal medicine and rheumatology at Johns Hopkins University, Baltimore. She is also a member of the ACR Insurance Subcommittee.
References
- Putman M. FDA approves nintedanib for SSc-ILD, but temper your expectations. The Rheumatologist. 2019 Dec 18.
- Kaufman M. FDA approves tocilizumab to treat systemic sclerosis-associated ILD. The Rheumatologist. 2021 Mar 25.
- Khanna D, Lescoat A, Roofeh D, et al. Systemic sclerosis–associated interstitial lung disease: How to incorporate two Food and Drug Administration–approved therapies in clinical practice. Arthritis Rheumatol. 2022 Jan;74(1):13–27.
- Denton CP, Khanna D. Systemic sclerosis. Lancet. 2017 Oct 7;390(10103):1685–1699.
- Nihtyanova SI, Sari A, Harvey JC, et al. Using autoantibodies and cutaneous subset to develop outcome-based Disease classification in systemic sclerosis. Arthritis Rheumatol. 2020 Mar;72(3):465–476.
- Elhai M, Meune C, Boubaya M, et al. Mapping and predicting mortality from systemic sclerosis. Ann Rheum Dis. 2017 Nov;76(11):1897–1905.
- Bernstein EJ, Jaafar S, Assassi S, et al. Performance characteristics of pulmonary function tests for the detection of interstitial lung disease in adults with early diffuse cutaneous systemic sclerosis. Arthritis Rheumatol. 2020 Nov;72(11):1892–1896.
- Meyer OC, Fertig N, Lucas M, et al. Disease subsets, antinuclear antibody profile, and clinical features in 127 French and 247 U.S. adult patients with systemic sclerosis. J Rheumatol. 2007 Jan;34(1):104–109.
- Tashkin DP, Roth MD, Clements PJ, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): A randomised controlled, double-blind, parallel group trial. Lancet Respir Med. 2016 Sep;4(9):708–719.
- Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006 Jun 22;354(25):2655–2666.
- Distler O, Highland KB, Gahlemann M, et al. Nintedanib for systemic sclerosis-associated interstitial lung disease. N Engl J Med. 2019 Jun 27;380(26):2518–2528.
- Khanna D, Lin CJF, Furst DE, et al. Tocilizumab in systemic sclerosis: A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med. 2020 Oct;8(10):963–974.
- Roofeh D, Lin CJF, Goldin J, et al. Tocilizumab prevents progression of early systemic sclerosis–associated interstitial lung disease. Arthritis Rheumatol. 2021 Jul;73(7):1301–1310.
- Hoffmann-Vold AM, Maher TM, Philpot EE, et al. The identification and management of interstitial lung disease in systemic sclerosis: evidence-based European consensus statements. Lancet Rheumatol. 2020 Feb 1;2(2):E71–E83.
