ADOBE STOCK / JARUN011
Antiphospholipid syndrome (APS) is an acquired thromboinflammatory disease that can have severe, sometimes catastrophic, effects on patients and their families. Our modern understanding of APS began to emerge in the early 1980s. At that point, it was defined as a condition characterized by thrombotic episodes and/or pregnancy complications in the presence of antiphospholipid antibodies (aPL).1
Clinically, APS can present with a broad spectrum of disease manifestations, which complicate both its diagnosis and management. Beyond its well-known thrombotic and pregnancy-related complications, APS routinely presents with a range of additional clinical features, including hematologic derangements, cardiac valve damage, chronic nephropathy, cognitive dysfunction and livedo reticularis/racemosa.2
APS is most commonly a standalone autoimmune diagnosis. It is also routinely associated with other autoimmune diseases, particularly systemic lupus erythematosus (SLE), in which case the APS diagnosis has historically been designated as “secondary.”
The severest form of the condition is catastrophic antiphospholipid syndrome (CAPS), which involves the simultaneous thrombosis of microvascular beds throughout the body, leading to multi-organ failure and sometimes death.
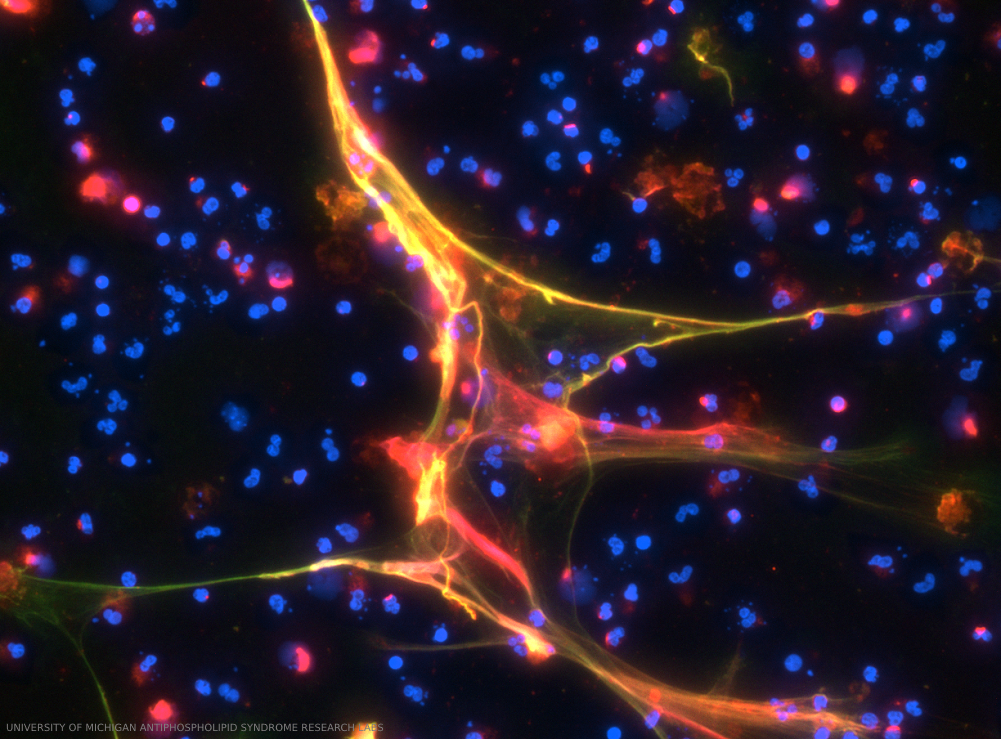
The figure illustrates the detection of anti-neutrophil extracellular trap (NET) antibodies from patients with primary antiphospholipid syndrome, by immunofluorescence microscopy. When NETs were incubated with plasma from patients with high levels of anti-NET IgG, antibodies robustly decorated NET strands. DNA stained blue, neutrophil elastase stained green and human IgG stained red. This is a merged image. Credit: UNIVERSITY OF MICHIGAN ANTIPHOSPHOLIPID SYNDROME RESEARCH LABS
Recent data have suggested the overall prevalence of APS is roughly 50 per 100,000 people, although more work on this front in diverse populations remains an area of need.2
What critical updates and practical insights should a rheumatology professional know about APS?
New ACR/EULAR APS Classification Criteria
The original classification criteria for APS, conceived in 1999 and revised in 2006, confirm the presence of disease when one or more aPL is persistently detectable in the circulation of an individual experiencing vascular thrombosis or obstetric morbidity. As of 2006, acceptable aPL tests included solid-phase immunoassays detecting the IgG and IgM isotypes of either anti-cardiolipin antibodies (aCL) or anti-beta-2 glycoprotein-I antibodies (aβ2GPI), as well as the lupus anticoagulant (LA) assay, which screens for aPL based on their paradoxical ability to prolong lab-based clotting times.
The criteria were a revelation for the field, but still fell short of fully capturing the systemic nature of APS. They also did not include a nuanced, risk-stratified approach that could account for other thrombotic risk factors. These gaps eventually sparked a transformative collaboration between the ACR and EULAR. Together, the groups crafted a multi-criteria additive points system for APS classification.
This modern approach marries expert insights with data-driven methods, resulting in a framework that spans six clinical domains denoted as venous thromboembolism, arterial thrombosis, microvascular, obstetric, cardiac valve and hematology—and two laboratory domains focused on aPL tests.3 The scoring system, which assigns one to seven points across each of these eight domains, allows for nuanced classification. A minimum of three points from the six clinical domains and three points from the two laboratory domains are now required to classify APS.3
These updated criteria herald at least three significant advancements. First, they more comprehensively account for the clinical heterogeneity of APS. Second, they allow additional risk factors to be taken into account when classifying patients. Finally, they more finely tune the expected impact of different aPL profiles on an individual’s risk.
It must be acknowledged, however, that the quest for high specificity in identifying uniform APS patient populations for research has led to a decrease in sensitivity—from 99% with the old criteria to 84% with the new.3 The clinical domains are also necessarily demanding, requiring detailed information for accurate classification.
 In 1994, antiphospholipid syndrome was named Hughes syndrome after Graham R.V. Hughes, MD, FRCP, a consultant rheumatologist and head of the London Lupus Centre. Check out his 2016 article on the condition here.
In 1994, antiphospholipid syndrome was named Hughes syndrome after Graham R.V. Hughes, MD, FRCP, a consultant rheumatologist and head of the London Lupus Centre. Check out his 2016 article on the condition here.Moreover, the criteria’s focus on solid-phase ELISA-based testing platforms, which was justified by their more reliable quantitative data, complicates the day-to-day implementation of these rules given that the majority of testing centers have already moved toward automated chemiluminescence- and fluorescence-based immunoassays. It should also be noted that the updated criteria de-prioritize the IgM isotypes of aCL and aβ2GPI, while emphasizing that more work is needed to understand the independent role of such antibodies in APS’s various clinical features.
In this new era, the clinician is reminded that the classification criteria were explicitly designed to optimally support research, not necessarily diagnosis. The latter will continue to require experience, nuance and judgment by the clinician, who must continue to weigh clinical manifestations against aPL profiles and other potential risk factors. Diagnosis will always remain something of an art form not fully captured (at least for now) by a computer model or a standardized algorithm. While classification criteria can offer insightful guidance, they must be applied with discernment and should never replace the personalized assessment necessary for accurate diagnosis.
The Pathogenesis of APS
One recent advancement in our understanding of APS has spotlighted the involvement of neutrophils and their inflammatory byproducts, known as neutrophil extracellular traps (NETs).4 Neutrophils are, of course, instrumental to the body’s innate defense against pathogens. In pursuit of containing pathogens, neutrophils release NETs, spider web-like structures built of nucleus-derived chromatin that becomes decorated with various antimicrobial enzymes. NETs are triggered not only by infections, but also by sterile stimuli, such as immune complexes and other inflammatory cues, and it is from this sterile perspective that they appear to contribute to the coagulopathy and inflammation inherent to APS.
Several decades ago, it was shown that aPL can activate neutrophils to release damaging reactive oxygen species and to express tissue factor on the cell’s surface. Elegant work between 2000 and 2010 found that neutrophils play an instrumental role in driving the dysfunctional placentation that is unfortunately common in pregnant individuals with APS. Most recently, it has been revealed that at least some aPL also triggers NET release, with these prothrombotic structures then able to engage with, and activate, clotting factors, endothelial cells, platelets and the complement system.4,5
Beyond the increased production of NETs, serum isolated from some patients with APS does not effectively degrade prothrombotic NETs. This may result in a vicious cycle of thrombo-inflammation, including complement system activation. Potentially adding further fuel to the fire, multiple groups have now shown that the key APS autoantigen β2GPI interacts not only with phospholipids, but also with the DNA-based backbone of NETs. Disrupted regulation of NETs is most likely to be appreciated in subgroups of patients with high-risk antibody profiles, recurrent thrombotic events and/or microvascular disease, all groups for which more effective treatments are needed. With an eye to the future, a 2024 study has identified circulating calprotectin, a relatively stable marker of NET release, as a promising biomarker that may eventually find value in the clinic.6
This burgeoning knowledge highlights a complex interaction between innate immunity and coagulation in APS that will, hopefully, pave the way for novel research and, most importantly, more effective therapeutic strategies for those grappling with this multifaceted disease.
Treatment Updates
Primary thrombosis prophylaxis is often considered for patients with a clinically significant aPL profile without a history of thrombosis.2 Studies show an annual thrombotic risk of 1.3–5.3% among aPL carriers, with individuals displaying triple positivity (defined by the presence of LA, aCL IgG, and aβ2GPI IgG) at the higher end of that range.
Preventive strategies should consider individual risk factors and the aPL profile. Low-dose aspirin has been explored for primary prevention in aPL-positive individuals, with mixed evidence regarding its efficacy. Although one prospective study with few events in either group found no significant benefit, meta-analyses and EULAR guidelines suggest potential protective effects, especially in the setting of high-risk aPL profiles or SLE, acknowledging that any decision must be balanced against an individual’s bleeding risk.
Statins may offer some protection against thrombosis by reducing inflammation and atherosclerosis, although their potential role in aPL-positive individuals without dyslipidemia is still not well defined.
Hydroxychloroquine has demonstrated promise in preventing thrombosis in patients with SLE and aPL-positive carriers. Despite limited prospective trial data in APS specifically, its use may be considered in selected individuals, for example, those with high-risk aPL profiles and some features of SLE.
For patients with APS and a history of prior thrombotic events, warfarin remains the preferred treatment.2,5 In the general population, newer, direct oral anticoagulants (DOACs) offer obvious benefits over warfarin, including no need for routine monitoring, fewer drug interactions and no dietary restrictions. However, prospective trials have raised concerns about the use of DOACs in individuals with APS, with a particular concern for an increased risk of breakthrough arterial thrombotic events. Some international guidelines (2019 European Society of Cardiology, American Society of Hematology) advise against DOACs for all APS patients, while others (EULAR, British Society for Haematology, International Society on Thrombosis and Haemostasis) suggest DOACs may be considered for certain patients with lower-risk aPL profiles and a history of venous, but not arterial, thrombosis.2
We support a tailored approach to secondary thrombosis prophylaxis based on an individual patient’s circumstances. Although warfarin should remain the first-choice agent, DOACs could be considered for non-triple-positive patients who have shown many years of stability on a DOAC, who are unwilling to undergo INR monitoring, who have poor adherence to warfarin or who have a history of serious adverse events or contraindications to warfarin.
Treatments on the Horizon
Current management of APS primarily focuses on risk modification and anticoagulation, but advancements in our understanding of the pathogenesis of APS have revealed new potential therapeutic targets in the immune system. A review of clinicaltrials.gov finds active trials focused on new approaches to anticoagulation (e.g., higher-dose DOACs), immunomodulation (e.g., hydroxychloroquine) and specific B cell antagonism (e.g., belimumab). Other pathways, including complement activation and NET release, likely also deserve further consideration.2 A few examples of ongoing efforts are discussed next.
TNF Inhibitors for Obstetric APS: Evidence from animal models has identified a promising role for anti-tumor necrosis factor α (TNF-α) agents in improving APS-associated pregnancy outcomes. A small, prospective study reported positive results in 70% of women treated with adalimumab or certolizumab, supporting further exploration of TNF inhibitors in obstetric APS management. An ongoing, open-label study of certolizumab in pregnant individuals with APS may add additional clarity to this topic.
Daratumumab (Anti-CD38): Routinely used in multiple myeloma, the plasma cell-depleting agent daratumumab has recently been considered as a potential induction treatment for APS, with the goal of resetting humoral immunity. A single case report suggested benefits in a patient with refractory APS, and an ongoing trial is now systematically assessing its safety and potential efficacy in decreasing aPL levels in individuals with triple-positive APS.
Chimeric Antigen Receptor (CAR) T Cell Therapy: This innovative approach to immunotherapy modifies autologous or allogeneic T cells to kill pathogenic cells in the host. One report of CD19-targeting CAR-T cell therapy found a reduction in aCL IgM levels in a patient with refractory APS and lymphoma with CAR-T treatment. Additionally, intriguing preclinical studies have described next-generation cellular therapies that might be able to specifically identify and then kill anti-β2GPI-expressing B cells, offering a glimpse of the type of precision medicine that may be on the horizon for diseases like APS.
Summary
Although numerous challenges persist, the new classification criteria mark a significant advance in acknowledging APS’s complexity and systemic nature. As our grasp of APS’s underlying pathogenesis and molecular dynamics deepens, one hopes new therapeutic avenues will emerge that are both more personalized and more precise.
It has been more than three centuries since Queen Anne’s lupus and APS left their mark on our history (see sidebar, right) and more than four decades since APS began to be systematically defined. Yet awareness of APS remains sparse in many regions across the U.S. and the world, leaving significant gaps in care and information for many of those affected by the condition.
 At Michigan’s APS Program, we are working to bridge this gap through our research and by offering patient-centric information and practical advice to enhance the quality of life of those living with APS. Our approach emphasizes collaboration with our patients, involving them directly in our initiatives as we together focus on developing solutions centered on their needs. Patient engagement in APS research is likely to not only enhance the relevance and effectiveness of individual studies, but also to lead to types of scientific advancements that will meaningfully improve patient outcomes. Please refer us your patients (scan or click the code).
At Michigan’s APS Program, we are working to bridge this gap through our research and by offering patient-centric information and practical advice to enhance the quality of life of those living with APS. Our approach emphasizes collaboration with our patients, involving them directly in our initiatives as we together focus on developing solutions centered on their needs. Patient engagement in APS research is likely to not only enhance the relevance and effectiveness of individual studies, but also to lead to types of scientific advancements that will meaningfully improve patient outcomes. Please refer us your patients (scan or click the code).
Yu Zuo, MD, MS, is a rheumatologist at the University of Michigan Medical Center, Ann Arbor.
Jason S. Knight, MD, PhD, is a rheumatologist at the University of Michigan Medical Center, Ann Arbor.
References
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006 Feb;4(2):295–306.
- Ambati A, Knight JS, Zuo Y. Antiphospholipid syndrome management: A 2023 update and practical algorithm-based approach. Curr Opin Rheumatol. 2023 May;35(3):149–160.
- Barbhaiya M, Zuily S, Naden R, et al. The 2023 ACR/EULAR antiphospholipid syndrome classification criteria. Arthritis Rheumatol. 2023 Oct;75(10):1687–1702.
- Ambati A, Zuo Y, Knight JS. An update on inflammation in antiphospholipid syndrome. Curr Opin Rheumatol. 2023 Mar;35(2):89–97.
- Knight JS, Branch DW, Ortel TL. Antiphospholipid syndrome: Advances in diagnosis, pathogenesis, and management. BMJ. 2023 Feb;380:e069717.
- Hoy CK, NaveenKumar SK, Navaz SA, et al. Calprotectin impairs platelet survival in patients with primary antiphospholipid syndrome. Arthritis Rheumatol. 2024 Jan 16.
- Tincani A, Fontana G, Mackworth-Young C. The history of antiphospholipid syndrome. Reumatismo. 2023 Mar;74(4).
