“The ACR and CHEST have released two new guidelines for the screening, monitoring and treatment of patients with interstitial lung disease, and our article summarizing the recommendations is perfect for the busy clinician,” says Dr. Bharat Kumar, physician editor of The Rheumatologist.

 Working in conjunction with the American College of Chest Physicians, the ACR recently released two new guidelines on the screening, monitoring and treatment of patients with interstitial lung disease (ILD) secondary to systemic autoimmune rheumatic diseases (SARDs). The guidelines will help clinicians quickly diagnose and optimally manage these diseases.1,2
Working in conjunction with the American College of Chest Physicians, the ACR recently released two new guidelines on the screening, monitoring and treatment of patients with interstitial lung disease (ILD) secondary to systemic autoimmune rheumatic diseases (SARDs). The guidelines will help clinicians quickly diagnose and optimally manage these diseases.1,2
Background

Dr. Sindhu Johnson
Over 50% of patients with systemic sclerosis develop ILD, and rheumatologists have long recognized this major cause of morbidity and mortality.1 Sindhu R. Johnson, MD, PhD, professor of medicine at the University of Toronto, Canada, director of the Toronto Scleroderma Program and principal investigator for the guideline, notes that more recent studies have increased our awareness of the risks for ILD in other SARDs, such as rheumatoid arthritis and Sjögren’s disease. These have comparatively lower, but still significant, disease incidence.
When it occurs in the context of a SARD, progression of ILD varies widely. It can present with subclinical features, such as mild cough, following a slow, progressive course, or with more acute manifestations and rapid disease progression with severe deterioration of pulmonary function. Alveolitis from activated T cells can result in the release of cytokines and the accumulation of alveolar macrophages, T lymphocytes and neutrophils in the lungs. Ongoing inflammation can lead to tissue injury and, sometimes, eventually fibrosis, which may be progressive.3
Dr. Johnson points out that the number of immunosuppressants and other therapies available to treat ILD has expanded in recent years. In idiopathic (non-SARD-related) ILD, antifibrotics, such as nintedanib or pirfenidone, are often first-line therapy.

Dr. Elana Bernstein
But Elana J. Bernstein, MD, MSc, Florence Irving associate professor of medicine in the Division of Rheumatology at Columbia University, New York City, and co-first author of the guideline, points out that immunosuppressants are the cornerstone of therapy for SARD-ILD because they address ongoing inflammation.Like other recent ACR guidelines, these rely on the GRADE methodology (Grading of Recommendations, Assessment, Development and Evaluation) to rate the certainty of evidence on the basis of a literature review targeted to specific, clinically relevant PICO (i.e., population, intervention, comparator and outcome) questions. A panel that included both clinicians and patients voted on the final recommendations. Dr. Bernstein underscores that these recommendations are meant to guide shared decision making between clinicians and patients, and should not be used by third parties to constrain diagnostic or treatment options.
These guidelines and recommendations apply specifically to adult patients with systemic sclerosis, idiopathic inflammatory myopathies, mixed connective tissue disease, rheumatoid arthritis or Sjögren’s disease.
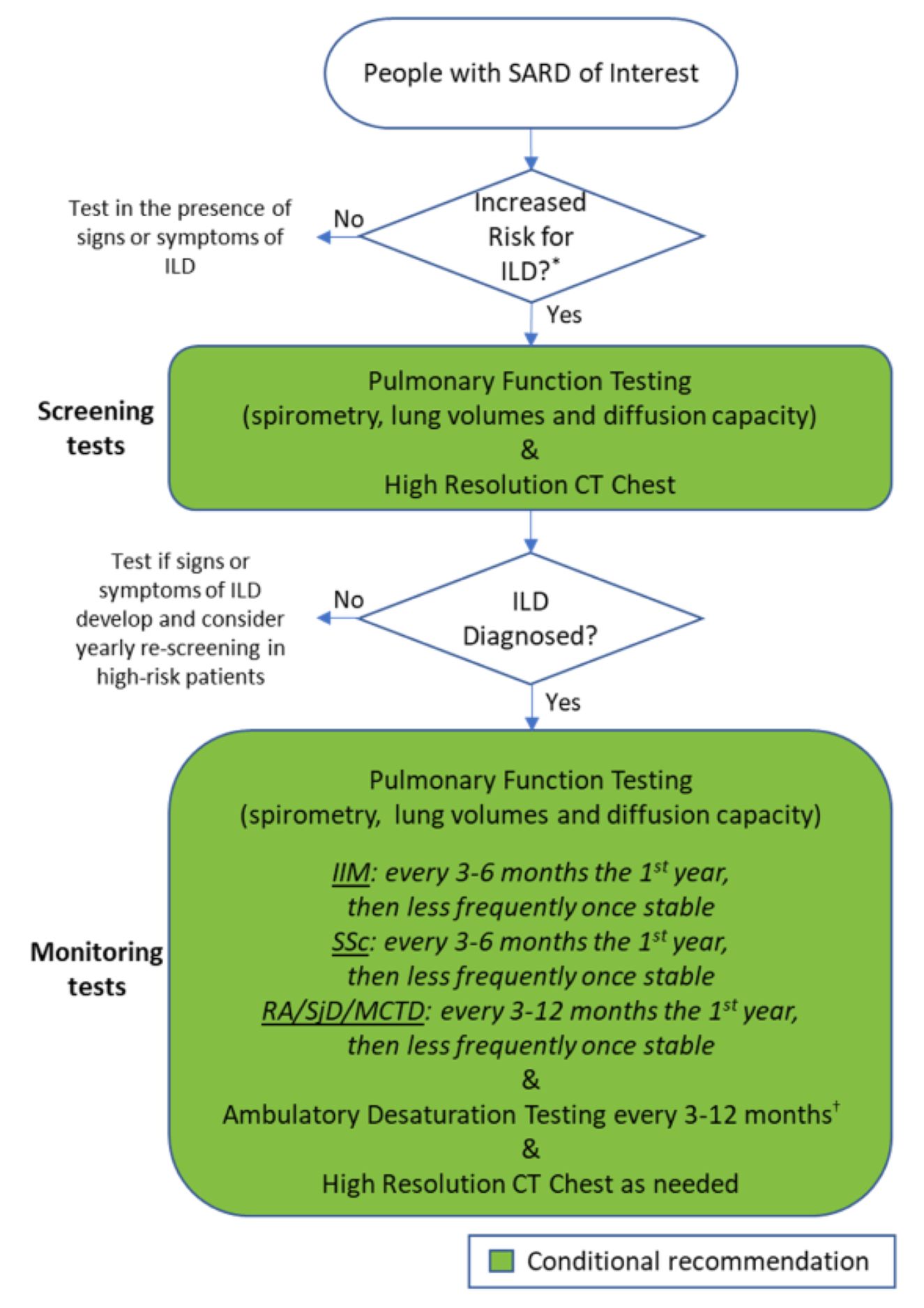
Figure 1: Recommendations for Testing. (CLICK TO ENLARGE)
Screening & Monitoring for ILD
To help with screening decisions, the first guideline provides a detailed list of disease-specific factors that may increase a patient’s risk of developing ILD, drawing on elements from a patient’s medical history, current presentation and laboratory results.1 Patients who lack the risk factors and are asymptomatic may not necessarily need screening beyond their regular history and physical exam.
However, Dr. Johnson notes that clinicians should still discuss the possibility of screening with such patients. “If clinicians are uncertain whether a patient should be screened or not, they should err on the side of caution and screen,” she says.
Recommendations: Pulmonary function tests (PFTs) are conditionally recommended over history and physical exam or ambulatory desaturation testing alone to screen for ILD in SARD patients at higher risk.
Others conditional recommendations are for high-resolution computed tomography (HRCT) of the chest over PFTs alone, and for HRCT over history and physical exam, as well as in favor of chest HRCT with PFTs over PFTs alone.
Although not enough evidence was available for specific recommendations, the guideline provides general guidance that most people with systemic sclerosis, idiopathic inflammatory myopathies or mixed connective tissue disease with features of systemic sclerosis should have ILD screening tests at presentation, as should many patients with rheumatoid arthritis or Sjögren’s disease who have risk factors.
Dr. Bernstein shares that her strong preference is to screen all people with systemic sclerosis with HRCT at presentation, due to the high prevalence of the disease in this population and the fact that PFTs alone may miss early ILD.
Dr. Johnson also points out that office spirometry with tests of forced vital capacity or forced expiratory volume in one second are not sufficient for screening; screening PFTs must also include total lung capacity and diffusion capacity. Additionally, she adds that a CT angiogram, if incidentally ordered for other indications, is not sufficiently sensitive to screen for ILD.
The Patient Panel expressed a strong preference for early diagnosis of ILD, even if more frequent screening leads to identification of subclinical disease or incidental findings that may require further testing.
Once diagnosed with ILD, SARD patients require monitoring for their response to therapy, typically in conjunction with pulmonology.
Recommendations: In SARD patients diagnosed with ILD, conditional recommendations are for monitoring with PFTs and HRCT over PFTs alone. An additional conditional recommendation is for added monitoring with ambulatory desaturation testing.
Rheumatologists are familiar with PFTs and HRCT, but “they may be less familiar with ambulatory desaturation testing,” says Dr. Johnson. “This recommendation is a great example in which co-management with pulmonologists is valuable [because] ambulatory desaturation testing is part of their scope of practice.”
The guideline also provides general suggestions based on expert opinion and patient preferences on potential frequency of these tests in specific contexts, which can be adapted to patients’ individual needs. For example, patients with systemic sclerosis may initially be monitored with PFTs every three to six months, ambulatory desaturation testing every three to 12 months and HRCT when clinically indicated. The Patient Panel strongly preferred more frequent testing over less frequent testing to avoid missing signs of progression.
Treatment of ILD in SARDs
Most recommendations in the treatment guideline were based on low or very low certainty of evidence, as were the recommendations on screening and monitoring treatment. Dr. Johnson points out that in these circumstances, patients’ preferences and values more strongly influence treatment choice. “The Patient Panel told us they wanted clear communication about the advantages and side effects [of therapies], so they could be actively involved in their choice of medication,” she says.
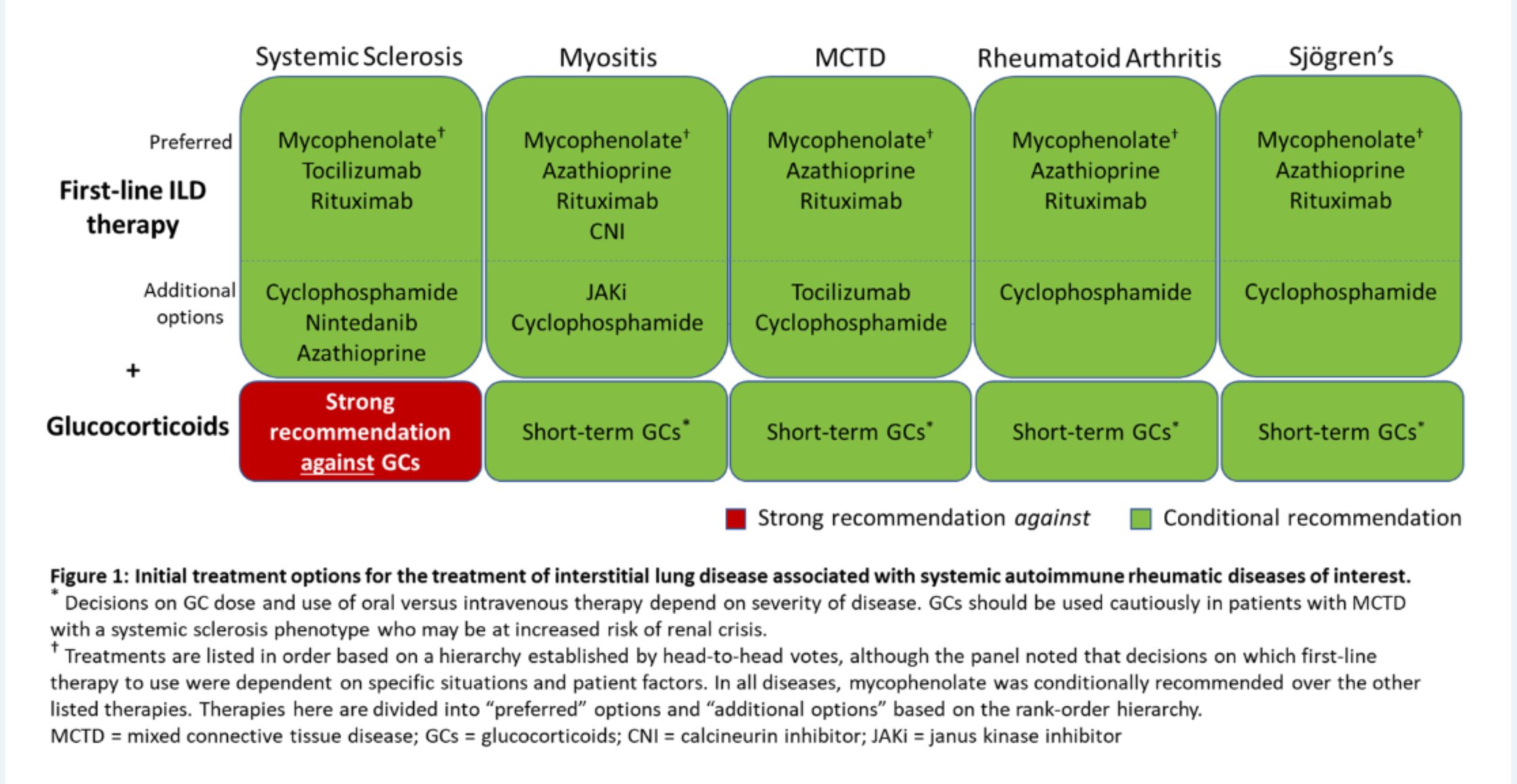
Figure 2: Initial Options for the Treatment of ILD Associated with Systemic Autoimmune Rheumatic Diseases. (CLICK TO ENLARGE)
The guideline recommendations are stratified by first-line treatments versus treatments to consider if a patient’s disease has progressed after initial treatment, with certain recommendations specific to individual SARDs. Additional recommendations clarify additional treatment approaches in patients with rapidly progressing disease.
Glucocorticoids
Recommendations: A strong recommendation against glucocorticoid use in patients with systemic sclerosis as first-line treatment of SARD-ILD was made. However, for patients with ILD related to other SARDs, the conditional recommendation is in favor of glucocorticoid treatment in conjunction with another therapy, usually a traditional immunosuppressive.
The strong recommendation against glucocorticoids as first-line ILD treatment in systemic sclerosis, one of the few strong recommendations in the guideline, is due to the risk of inducing renal crisis in this population. “I think rheumatologists are very familiar with this risk, but people outside the rheumatology community [may] be less so,” says Dr. Bernstein.
In all likelihood notes Dr. Johnson, clinicians should also avoid glucocorticoids in patients with mixed connective tissue disease who have a scleroderma-dominant phenotype.
Recommendations: Long-term glucocorticoid use is strongly recommended against in systemic sclerosis and conditionally recommended against in other types of SARD-ILD in patients with progression after initial first-line therapy.
Although these agents should not be used for long-term treatment in any SARD-ILD due to side effects and low evidence of efficacy, Dr. Bernstein points out that short-term treatment with glucocorticoids may sometimes be appropriate (e.g., in a flaring patient or as a bridge to another therapy).
Immunomodulators: Initial Treatment
Recommendations: Mycophenolate, rituximab, cyclophosphamide and azathioprine are conditionally recommended as first-line treatments for SARD-ILD, with mycophenolate conditionally recommended above these other agents.
Dr. Johnson notes that guideline developers preferred some agents due to published literature and expert experience, but other agents may be options in specific circumstances. Thus, although these are all potential first-line options, cyclophosphamide and azathioprine may be considered additional options rather than preferred first-line treatment.
Comorbidities or extrapulmonary disease may also influence treatment choice (e.g., rituximab may be used in a patient with rheumatoid arthritis and ILD to address both conditions).
Dr. Bernstein notes that many physicians may be reluctant to prescribe cyclophosphamide due to concerns of infection, hemorrhagic cystitis and potential infertility; mycophenolate has a comparatively better side effect profile.
Due to evidence in specific SARD-ILD populations, some specific immunomodulators are recommended for or against in specific SARD groups. These are not necessarily preferable to other treatment options listed above, but can provide additional options for these specific groups.
Recommendations: Tocilizumab is conditionally recommended as a potential first-line option specifically in the context of systemic sclerosis (although mycophenolate may be preferred), and calcineurin inhibitors or JAK inhibitors may be considered as additional options for inflammatory myositis. (For additional examples on medications and specific conditions, see the full guideline.)
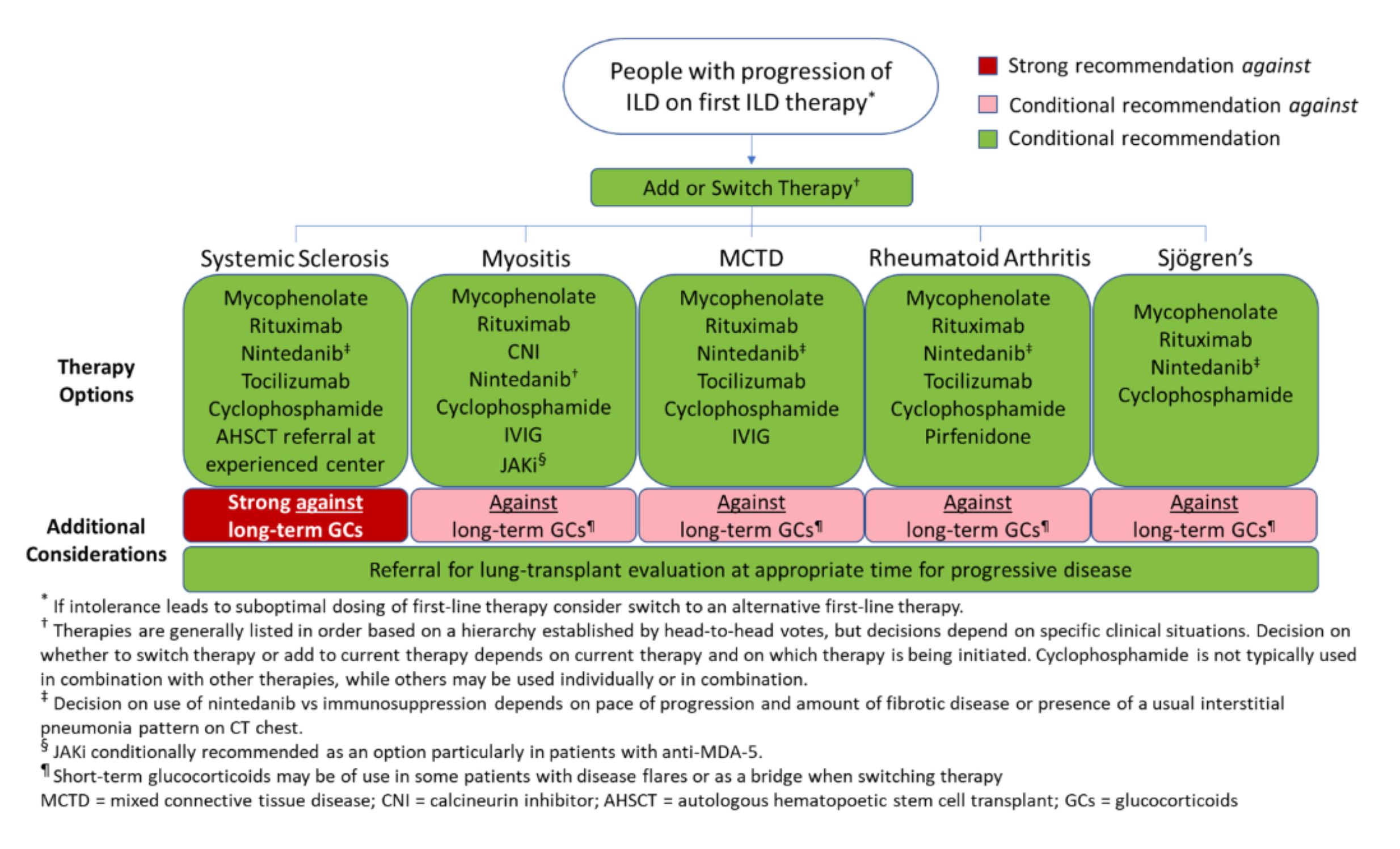
Figure 3: ILD Progression Despite Therapy. (CLICK TO ENLARGE)
Immunomodulators: Treatment After Progression
Recommendations: The conditional recommendation for progression of SARD-ILD after initial treatment is to add or switch therapies to mycophenolate, rituximab, cyclophosphamide or nintedanib (discussed below). If not initially chosen as first-line therapy, mycophenolate is preferred.
For treatment in the event of ILD progression after initial treatment, the guideline provides a list of ranked therapies to consider in specific SARD-ILDs based on head-to-head votes. In addition to the therapies just listed, therapies to consider in specific SARD-ILDs may include pirfenidone, tocilizumab, Janus kinase (JAK) inhibitors, calcineurin inhibitors, intravenous immunoglobulin (IVIG) and/or referral for hematopoietic stem cell transplant or lung transplant at experienced centers.
Additional Therapies: Antifibrotics
With the emergence of antifibrotics as treatment options, some clinicians have been unclear about the best ways to employ them with immunosuppressants.
Recommendations: For people with SARD-ILD, the conditional recommendation is against initially combining an antifibrotic with mycophenolate and against adding an antifibrotic to mycophenolate in the absence of ILD progression.
Although nintedanib can be considered as a potential additional option in first-line treatment for patients with systemic sclerosis, Dr. Bernstein notes that she would very rarely consider it a first-line choice, and it should primarily be used in the context of progressive disease. Another antifibrotic, pirfenidone, may also be considered for progressive ILD in rheumatoid arthritis.
Nintedanib frequently causes gastrointestinal side effects, such as diarrhea. Especially in the systemic sclerosis population already at risk for such problems, it may not be the best option. Additionally, studies haven’t shown that nintedanib stabilizes or improves pulmonary forced vital capacity, although it may slow its decline.2
Dr. Johnson shares that pulmonologists often have more experience prescribing antifibrotics compared with immunosuppressives. But the rheumatology perspective can help in this situation because, in many cases, adding or switching to a different immunosuppressant may be more appropriate than an antifibrotic.
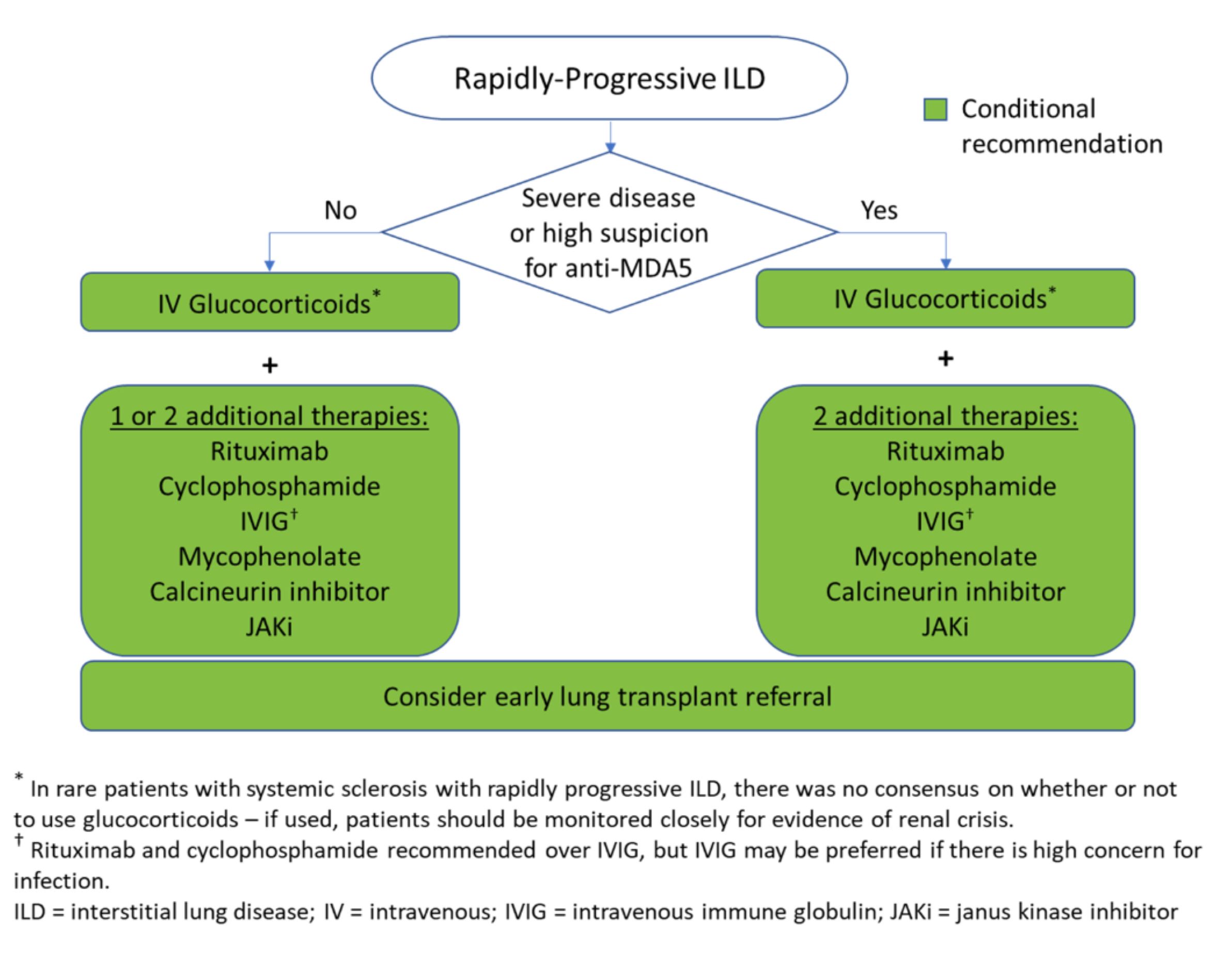
Figure 4: Rapidly Progressive ILD. (CLICK TO ENLARGE)
Additional Recommendations
The guidelines also contain recommendations on the management of rapidly progressing ILD, an aggressive and rare manifestation that occurs most often in the setting of idiopathic inflammatory myopathy. Patients positive for the anti-MDA-5 antibody may need particularly aggressive treatment, but this information may not always be available acutely.
Dr. Bernstein says it’s key to initially treat aggressively in any patient with a severe presentation. “Up front, you may use a combination of three immunosuppressant medications in a patient with severe disease,” she notes. “Then, once the patient stabilizes, you can start to peel back therapy.”
Dr. Johnson explains that some centers are opting for IVIG (with intravenous methylprednisolone) as a potential treatment option over rituximab or cyclophosphamide with intravenous methylprednisolone for patients with rapidly progressing disease due to the concern for infection in these critically ill patients. However, she notes that medical practice is changing in advance of the scientific literature; little published data directly support this approach. For additional insight on the management of rapidly progressing disease and other treatment, screening and monitoring recommendations, see the full guidelines.
Navigating different treatment options in individual SARD-ILDs can be challenging, particularly for clinicians with less experience with this population. “Don’t hesitate to call a colleague at an expert center if you are uncertain about which therapies to use, especially if the patient has comorbidities or extrapulmonary considerations,” urges Dr. Johnson.
Ruth Jessen Hickman, MD, a graduate of the Indiana University School of Medicine, is a medical and science writer in Bloomington, Ind.
References
- Johnson SR, Bernstein EJ, Bolster MB, et al. 2023 American College of Rheumatology (ACR)/American College of Chest Physicians (CHEST) guideline for the screening and monitoring of interstitial lung disease in people with systemic autoimmune rheumatic diseases. Arthritis Rheumatol. 2024.
- Johnson SR, Bernstein EJ, Bolster MB, et al. 2023 American College of Rheumatology/American College of Chest Physicians (CHEST) guideline for the treatment of interstitial lung disease in people with systemic autoimmune rheumatic diseases. Arthritis Rheumatol. 2024.
- van den Bosch L, Luppi F, Ferrara G, Mura M. Immunomodulatory treatment of interstitial lung disease. Ther Adv Respir Dis. 2022;16:17534666221117002.


