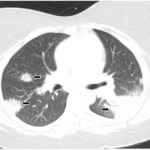
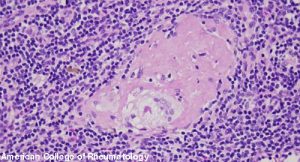
 BALTIMORE—Although first described by the English physician Sir Jonathan Hutchinson in the 1870s, sarcoidosis remains a prevalent—and insidious—disease in the modern world.
BALTIMORE—Although first described by the English physician Sir Jonathan Hutchinson in the 1870s, sarcoidosis remains a prevalent—and insidious—disease in the modern world.
At the 20th Annual Advances in the Diagnosis and Treatment of the Rheumatic Diseases Symposium at Johns Hopkins School of Medicine, Baltimore, Michelle Sharp, MD, MHS, assistant professor of medicine and co-director of the Johns Hopkins Sarcoidosis Program, provided a thorough lecture titled, Updates in Sarcoidosis.