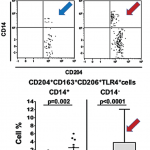
How macrophages (shown) behave when recognizing damage-associated molecular pathways can tell us why some inflammation doesn’t resolve, but becomes chronic and destructive.
royaltystockphoto.com/shutterstock.com
SAN FRANCISCO—To unravel how out-of-control inflammation begins in rheumatoid arthritis and other diseases, one target for immunologists is the macrophage. Researchers discussed macrophage activation and other key drivers of inflammation at the 2015 ACR/ARHP Annual Meeting on Nov. 7. How macrophages behave when recognizing damage-associated molecular pathways (DAMPs) tells us more about why some inflammation doesn’t resolve as it should, but instead becomes chronic and destructive.
Macrophage Polarization
Macrophages are involved in both adaptive and innate immunity. There appears to be some interaction or cross-talk between various cells that affect macrophage polarization, said Lionel B. Ivashkiv, MD, chief scientific officer of the Hospital for Special Surgery in New York.
In the past, macrophage polarization was viewed as taking one of two roads, based on what type of cytokine, such as tumor necrosis alpha or interferon gamma, the cells encountered. The two models were M1 or classical, which would trigger an inflammatory response, and M2 or alternative, which would normally lead to resolution of inflammation and tissue healing.2 In chronic inflammatory diseases, resolution doesn’t happen. RA synovial macrophages have more of a complex polarization spectrum, one where there may be a lot of overlap in gene expression, he said.3
Fibroblast-like synoviocytes modulate the expression of TNF-inducible genes in macrophages, he said. They also superinduce growth factor and prostaglandin pathways in macrophages.4
Synovial macrophages in RA are similar to TPP or C9 macrophages, which are also associated with chronic inflammation. “There’s also a striking similarity between the C9 polarization state and synovial fibroblast-trained macrophages,” he said. “What underlies this synergy?”
Fibroblasts and TNF cooperate to activate transcription factors in macrophages in a PG-dependent manner, and TNF and PGE2 also synergistically induce gene expression.5 Prostaglandins and growth factors also play a role in macrophage polarization. Synovial fibroblast-derived PGEs modulate macrophage TNF responses.
Fibroblast-like synoviocytes promote C9 macrophage polarization that may drive pannus formation and tissue damage in RA-affected joints, said Dr. Ivashkiv. More study of the C9 growth factor pathways may lead to better understanding of how this destruction happens in RA.
Macrophage Sub-Types
There’s little information about cell markers to identify subtypes of synovial macrophages, said Ursula Fearon, PhD, professor of molecular rheumatology at Trinity College in Dublin, Ireland.
In an RA synovium, you see more macrophages than normal. CD68 macrophages are increased in clinically inflamed knee joints, for example. “No matter the therapy, DAS 28 score changes significantly correlate to subsets of macrophages in the joints,” Dr. Fearon said.
Macrophages from the synovium of active RA joints have a pro-inflammatory profile, and there is an increase in CD50 and CD36.6 In RA synovial fluid, M1 phenotypes are seen in higher amounts. Anti-citrullinated protein antibodies (ACPA) also affect how macrophage subtypes are distributed in joints, which could lead to RA, she said.7 Dr. Fearon and colleagues studied the macrophage subtypes in synovial biopsies of very inflamed joints, and also saw an increase in M1 macrophages.
“It’s difficult to define the markers of macrophage subtypes,” said Dr. Fearon. As far as targeting macrophages to develop therapies, “many patients don’t respond to these targets.”
Researchers are focusing on reprogramming to try to block the metabolic pathway in macrophages, an old idea that has come back around, she said.
GLUT-1 mediated glucose metabolism drives a pro-inflammatory macrophage phenotype. Pyruvate kinase M2, a key metabolic regulator, tetramerization inhibits Hif-1α IL-1ß, and promotes an M2 phenotype.8 “So that’s a possible therapeutic target,” she said. Metformin has also been shown to directly suppress macrophage phenotypes. So synovial macrophages clearly play a role in RA disease activity, and one day may be targeted in the joint environment as a therapy, she said.
Turning Off Macrophages
“The function of a macrophage is to protect us from infection or injury,” said Andy Clark, PhD, professor of inflammation biology at the University of Birmingham in the United Kingdom. “When it detects one of these potentially harmful events, it responds by producing pro-inflammatory cytokines like TNF.”
Normally, this response is tightly regulated, he said. The macrophage returns to its resting state and stops producing TNF. However, “in the rheumatoid synovium, it looks as though macrophages have forgotten how to switch off. We need to understand how these on and off switches work, and why they sometimes don’t operate correctly.”
The key to understanding macrophages is the MAP-kinase (MAPK) p38 signaling pathway, said Dr. Clark. MAPK p38 inhibitors were discovered in a search for compounds that prevented macrophages from producing TNF. These compounds also reduced the expression of several other inflammatory factors. Although MAPK p38 inhibitors showed promise in experiments, these drugs haven’t proven to be effective at treating RA. That may be because we don’t yet completely understand how the MAPK p38 pathway works or how best to use these inhibitors, he said.
‘There’s also a striking similarity between the C9 polarization state & synovial fibroblast-trained macrophages. What underlies this synergy?’ —Lionel B. Ivashkiv, MD
The MAPK p38 signaling pathway exerts far-reaching control over inflammation, mainly by regulating the activity of the protein tristetraprolin (TTP), said Dr. Clark.
“We know that TTP is an anti-inflammatory protein because mice that don’t have TTP overproduce TNF and develop arthritis,” he said. “However, we can find lots of TTP in the joints of people with arthritis. For a long time, this has been puzzling. Now, we understand that TTP can be inactivated by MAPK p38. We think that the TTP we observe in rheumatoid joints must be inactivated, allowing TNF and other damaging factors to be overproduced.”

Dr. Clark
In one study, mice were genetically modified so that TTP could not be inactivated by MAPK p38, and these mice were dramatically protected against the development of arthritis, he said.9
The challenge now is to find a way to repair or reinforce the macrophage’s natural off switches, said Dr. Clark. One way to do this may be to reactivate dormant TTP in joints affected by rheumatoid arthritis. Clinical trials of MAPK p38 inhibitors have been disappointing, and interest in further studying these compounds has waned, he said. However, research to unravel how the MAPK p38 signaling pathway works is worth pursuing for future therapeutic targets. “In the future, drugs based on these compounds may be used to treat RA,” he said.
Susan Bernstein is a freelance medical journalist based in Atlanta.
References
- Donlin L, Jayatilleke A, Giannopoulou E, et al. Modulation of TNF-induced macrophage polarization by synovial fibroblasts. J Immunol. 2014 Sep 1;193(5):2373–2383.
- Murray P, Allen J, Biswas S, et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity. 21 Aug 2014;41(2):339–340.
- Ivashkiv L. Epigenetic regulation of macrophage polarization and function. Trends Immunol. 2013 May;34(5):216–223.
- Xue J, Schmidt S, Sander J, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014 Feb 20;40(2):274–288.
- Lee A, Qiao Y, Grigoriev G, et al. Tumor necrosis factor α induces sustained signaling and a prolonged and unremitting inflammatory response in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2013 Apr;65(4):928–938.
- Palacios B, Capetillo L, Izquierdo E, et al. Macrophages from the synovium of active rheumatoid arthritis exhibit an activin A-dependent pro-inflammatory profile. J Pathol. 2015 Feb;235(3):515–526.
- Zhu W, Li X, Fang S, et al. Anti-citrullinated protein antibodies induce macrophage subset disequilibrium in RA patients. Inflammation. 2015 Dec;38(6):2067–2075.
- Palsson-McDermott E, Curtis A, Goel G, et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1ß induction and is a critical determinant of the Warburg effect in LPS-activated macrophages. Cell Metab. 2015 Jan 6;21(1):65–80.
- Ross E, Smallie T, Ding Q, et al. Dominant suppression of inflammation via targeted mutation of the mRNA destabilizing protein tristetraprolin. J Immunol. 2015 Jul 1;195(1):265–276.
- Wilson-Gerwing T, Panahifar A, Cooper D, et al. Age-related differences in collagen-induced arthritis: Clinical, imaging, and biological characteristics in juvenile compared to adult animals (abstract). Arthritis Rheumatol. 2015;67(suppl 10).
- Inoue A, Matsumoto I, Tanaka Y, et al. TIARP attenuates auto-antibody mediated arthritis via the suppression of neutrophil infiltration into the joint (abstract). Arthritis Rheumatol. 2015;67(suppl 10).