The patient was evaluated by a dermatologist, who felt his cutaneous lesions were likely consistent with cutaneous PAN, which may be associated with group A Streptococcal infection. Differential diagnosis included panniculitis (including erythema nodosum, which may also be associated with group A Streptococcal infection, but would appear as septal panniculitis rather than medium vessel vasculitis; and erythema induratum, which may be associated with tuberculosis), ANCA-associated vasculitis, subcutaneous nodules of rheumatic fever and atypical infection. A repeat punch biopsy of the patient’s rash was performed.
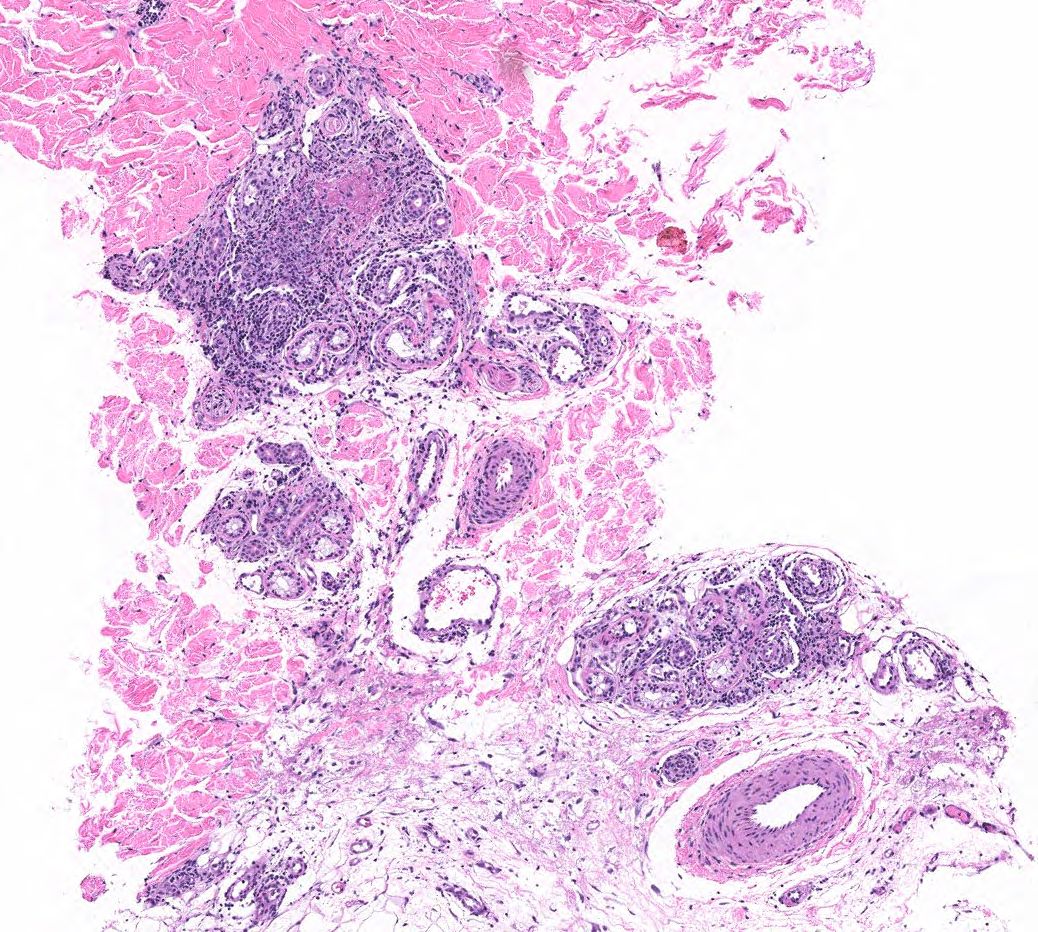
FIGURE 2B: Mid to deep dermal vasculocentric mixed inflammation with relatively unaffected deep dermal vessels (H&E stain, 10x). (Click to enlarge.)
The patient was started on treatment for rheumatic fever with 10 days of cephalexin because he met the 2015 American Heart Association revised Jones major criterion of subcutaneous nodules and minor criteria of polyarthralgia, fever of ≥38.5ºC and a CRP ≥3 mg/dL. He was also started on ibuprofen and colchicine for suspected cutaneous PAN. (Note: Penicillin was avoided due to the patient’s drug allergy.)
Discussion
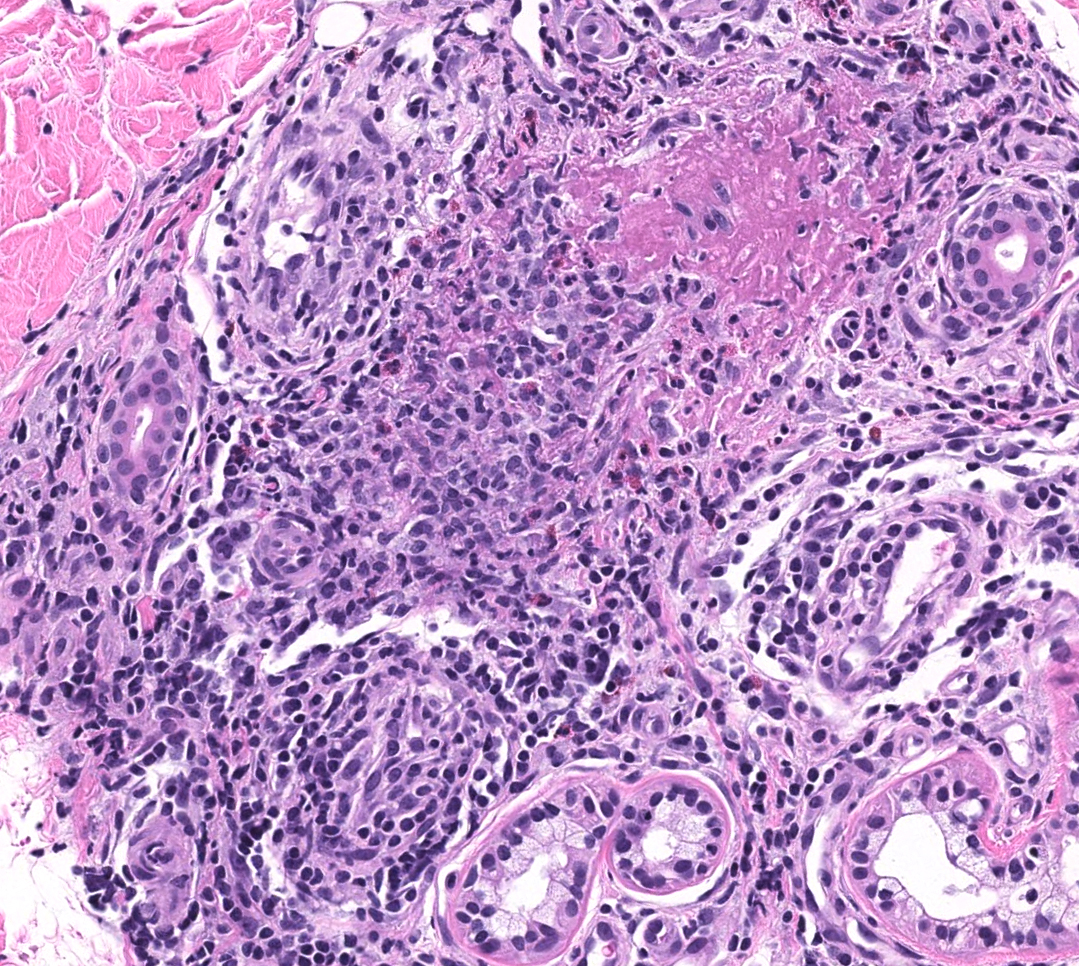
FIGURE 2C: Vasculocentric inflammation consisting of lymphocytes, histiocytes, neutrophils, and eosinophils with obscuration of vessel walls and fibrin deposition (H&E stain, 20x).
PAN is a rare, necrotizing, predominantly medium-vessel vasculitis first described in 1852, with its cutaneous-limited form described in 1931. Characteristic histopathology of cutaneous PAN is leukocytoclastic vasculitis in small- to medium-sized arterioles of deep dermis or hypodermis, with or without fibrinoid necrosis. Most cases of cutaneous PAN are idiopathic, but up to 40% may be associated with infection (group A Streptococcus, hepatitis B, hepatitis C, recurrent urinary tract infections, parvovirus B19 and Mycobacterium tuberculosis), as well as inflammatory bowel disease and long-term exposure to minocycline.5,6