The Mechanism of NSAID Cardiovascular Risk
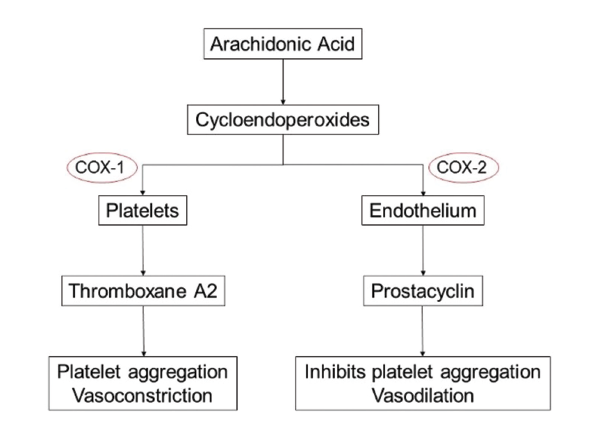
(click for larger image) Mechanism of Action
The relation of MI risk and COX-2 inhibition is most noted in a study conducted by Garcia-Rodriguez et al.7 They noted a direct correlation between MI risk and the degree of COX-2 over COX-1 inhibition. The exact mechanism of COX-2 inhibition remains unknown, but the hypothesis of an imbalance between thromboxane
A2 (which promotes platelet aggregation and acts as a vasoconstrictor) and prostacyclin (an inhibitor of platelet aggregation and a vasodilator), produced by both platelets and endothelial cells, has gained the most prominence.8-10
Similarly, it has been postulated that reduced prostaglandin synthesis due to NSAID use augments the Th-1 mediated immune response, which leads to increased proatherogenic cytokines. This ultimately leads to detrimental plaque remodeling, rupture and embolization of plaque.11
Researchers think the inhibition of prostaglandin synthesis increases peripheral vascular resistance and reduces renal perfusion, glomerular filtration and sodium excretion, which would ultimately lead to fluid retention and further contribute to the cardiovascular toxicity.11
More than 88,000 Americans suffered myocardial infarction due to rofecoxib, & more than 38,000 died.
Myocardial Infarction
The Downfall of COX-2 inhibitors Selective inhibitors of COX-2 came under closer scrutiny after two landmark trials, the Vioxx Gastrointestinal Outcomes Research study and the Adenomatous Polyp Prevention on Vioxx (APPROVe) studies.12,13 Both trials demonstrated an increased risk of cardiovascular events, which led to the withdrawal of rofecoxib from the market in 2004. It was subsequently estimated that more than 88,000 Americans suffered myocardial infarction due to this drug, and more than 38,000 died.