Over the past two decades, exciting research has provided an important new perspective on the biology of bone in health and disease, revealing an active and sometimes loud crosstalk between bone and the immune system. This perspective is a radical departure from the traditional view of the relationship of these tissues. In the traditional view, the bone and immune system represented separate functional realms despite their close physical relationship in the bone marrow. There, the bone houses immune cells and provides a sanctuary for key events in immune cell development and differentiation. Many of the important discoveries on the interactions between bone and the immune system arose from studies on arthritis, where immune cells and bone are closely juxtaposed in the joint microenvironment.1–4 The dimension and variety of these interactions, however, were unexpected and have spurred the creation of a dynamic new field of research.
From a clinical perspective examining pathogenesis, bone seems to be the target and victim from damage inflicted at the hands of the immune system. Indeed, in inflammatory arthritis, bone erosion is the dreaded outcome of the immune system’s destructiveness. As is now realized, however, this view is too simplistic because bone can drive the immune system as well as withstand its blows. Evidence for an active role of bone in the pathogenesis of autoimmune diseases, such as systemic lupus erythematosus and inflammatory arthritis, has accumulated rapidly. This evidence includes provocative new findings on the properties of bone marrow niches, which are specialized microenvironments for the maturation of immune cells and for the maintenance of immunological memory. This new research field is called osteoimmunology and addresses the mutual interactions of bone and the immune system.5
Although pioneering works in the late 1970s identified potential interactions between bone and the immune system, the field truly emerged from landmark studies in the late 1990s. These studies demonstrated that T lymphocytes can trigger bone loss by inducing differentiation of osteoclasts.4,6 In this article, we will discuss current concepts of osteoimmunology in the context of rheumatologic disease. We will focus on the processes of bone degradation and formation that are regulated by the immune system in inflammatory arthritides. We will also consider the role of bone and bone marrow as a niche for immune cells, a mostly unknown yet important facet for the interplay of bone and immune systems in the pathogenesis of autoimmune disease.
RANKL has been established as a key mediator of inflammatory bone loss and one of the key molecules in the interactions that characterize osteoimmunology.
Osteoclasts, Osteoblasts, and Inflammatory Cells
Bone is living tissue and not just an inert or rigid framework to organize other tissues in the body. Bone grows, heals, transforms, and constantly remodels as it adapts to external mechanical forces. Adult vertebrates replace approximately 10% of their total bone content per year, with adolescents showing even higher turnover rates. The cells mediating these dynamic processes are the osteoclasts that degrade bone and the osteoblasts that produce new bone. A tightly regulated balance of osteoclastic and osteoblastic activity maintains a steady state of total bone mass in healthy adults. Diseases affecting bone, however, can disturb this balance and lead to both the loss and gain of bone tissue.7
Immune-osteoclast Interactions
To remodel bone efficiently, osteoclasts and osteoblasts interact extensively; in fact, osteoblasts are important regulators of osteoclast differentiation. Elegant in vitro systems have demonstrated that bone marrow–derived monocytes/macrophages differentiate into osteoclasts in the presence of osteoblast precursors or other mesenchymal cells such as synovial fibroblasts.4 To induce osteoclastogenesis, osteoblast precursors and synovial fibroblasts release soluble factors, including macrophage colony–stimulating factor, and express surface molecules, such as the long-sought osteoclast differentiation factor (see Figure 1, p. 37).1,4 When cloned in 1998, the osteoclast differentiation factor surprisingly showed identity with a protein called the receptor activator of nuclear factor–κB ligand (RANKL).8,9 RANKL had already been known for its role in the immune system, particularly for lymph node development, demonstrating clearly shared signalling mechanisms between bone and the immune system.10
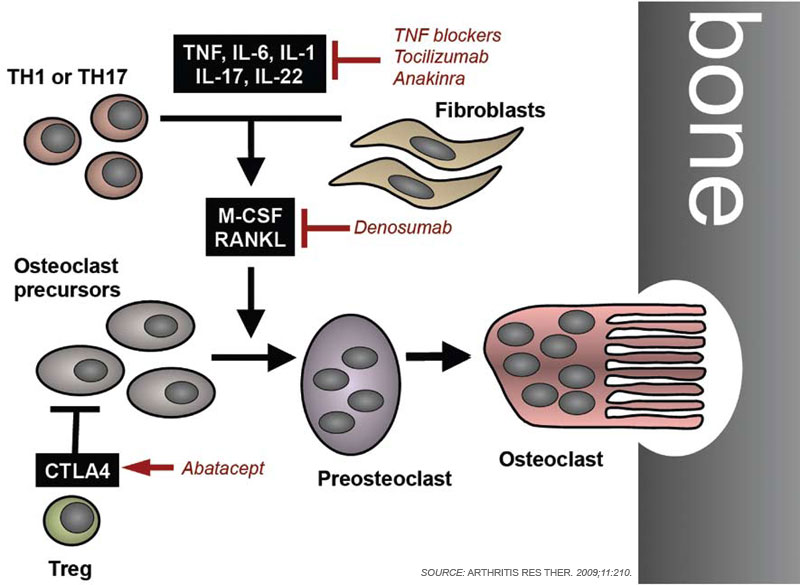
Since its discovery, RANKL has been established as a key mediator of inflammatory bone loss and one of the key molecules in the interactions that characterize osteoimmunology. When expressed on osteoblast precursors, synovial fibroblasts, and immune cells (see below), RANKL can bind to its receptor RANK on osteoclast precursors and induce osteoclastogenesis via intracellular signaling by NF-κB and the activation protein (AP)–1 transcription factor family.1,4 Studies from mouse models have provided important insights into the role of RANKL in bone. For example, mice deficient for RANKL show an osteopetrotic phenotype and a defective development of osteoclasts and low bone resorption. Interestingly, the expression of RANKL in the body is induced by proinflammatory cytokines, in particular tumor necrosis factor (TNF) as well as interleukin (IL)-1 and IL-17; this pattern of regulation further highlights the involvement of the immune system and inflammation in RANKL-mediated effects on bone.1,11
In addition to mesenchymal cells, inflammatory cells—T lymphocytes in particular—are key elements that regulate osteoclastogenesis. Clinical observations suggest that osteoclastic bone resorption is associated with T lymphocyte activation in autoimmune and inflammatory disease. The role of T cells in osteoclastogenesis and bone metabolism is further supported by findings indicating that activated T lymphocytes express high levels of RANKL. The actual effect of T lymphocytes on bone, however, is modulated by their cytokine expression repertoire and their pattern of surface molecule expression.
Although T cells can stimulate osteoclastogenesis, these cells also can display antiosteoclastogenic effects despite the expression of RANKL. This effect occurs particularly with TH1 cells expressing interferon-γ, a potent antiosteoclastogenic cytokine, and with regulatory T cells, which express high levels of CTLA4, a surface molecule important in inhibition of co-stimulation. In contrast, TH17 cells releasing IL-17 as well as TNF have been identified as important inducers of osteoclast differentiation.12 In this context, IL-17 may act in two ways. First, IL-17 can induce RANKL expression on mesenchymal cells; second, this cytokine causes local inflammation by the release of TNF, IL-17, and IL-22, further supporting RANKL expression.2,4
Immune-osteoblast Interactions
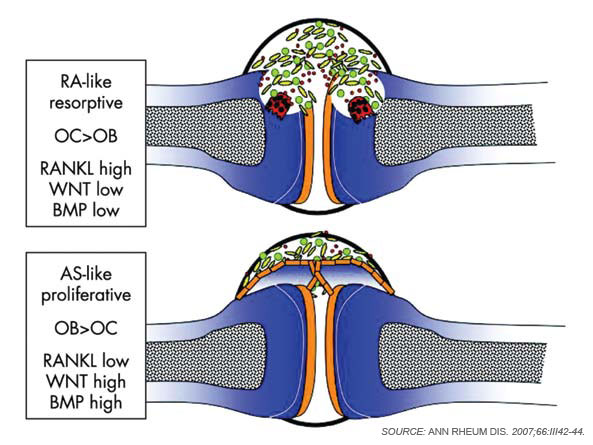
Although osteoclasts have been a major focus of attention in the study of inflammatory bone loss, surprisingly little is known about the activity of osteoblasts in this setting. Nevertheless, because bone metabolism depends on the balance between bone-resorbing osteoclasts and bone-forming osteoblasts, reduced bone formation likely contributes to bone loss in patients with arthritis. The mechanism behind this interaction is not fully defined, although TNF and other factors may repress bone formation, at least in part through the induction of dickkopf (Dkk)-1 and sclerostin, both inhibitors of the Wnt signaling, which is a central pathway for bone formation.13 Whereas Dkk-1 and sclerostin may act to suppress bone responses in diseases like rheumatoid arthritis (RA), impaired expression of these molecules also may be linked to aberrant bone formation, as found in spondyloarthritis (SpA). In addition to the effects of Wnt signaling, bone morphogenic proteins and transforming growth factor-β may induce new bone formation in arthritis.14 (See Figure 2, p. 37.)
Bone Erosion in RA
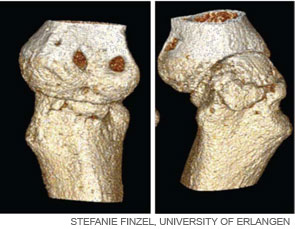
Inflammation is the primary trigger for the process of local bone erosions in RA, consistent with the prominence of inflammatory cell infiltrates in the tissue of patients with this disease (see Figure 3, at right). Bromley and Woolley and Gravallese and colleagues provided the first detailed descriptions of the location and activity of osteoclasts in the RA joint.15,16 These investigators demonstrated the expression of osteoclasts at the interface between advancing pannus tissue and the bone surface. Later, several groups demonstrated the essential role of osteoclasts in triggering inflammatory bone erosions in experimental disease models by blocking essential molecules for osteoclastogenesis or studying osteoclast-deficient mice.2,4
RANKL as well as M-CSF, the key mediators of osteoclastogenesis, show prominent expression in the synovium of patients with RA; this expression, however, does not appear to be balanced by the expression of regulatory molecules such as RANKL’s decoy receptor osteoprotegerin.2 In addition, inflammatory cytokines that induce RANKL, such as IL-1, IL-6, and TNF, are abundantly expressed in the synovial tissue of RA.4 The strong expression of RANKL in inflamed RA joints makes this molecule a potential important target for novel antiresorptive therapy. Evidence of this possibility comes from studies showing that RANKL-deficient mice are resistant to inflammation-induced bone loss but nevertheless show similar level of inflammation as their wild-type littermates.2,4
In humans, blockade of RANKL with the humanized monoclonal antibody denosumab is effective not only in the treatment of postmenopausal osteoporosis and bone loss from cancer metastasis but also in inhibiting bone destruction in RA.17–19 The strong effects of denosumab on erosion in RA are remarkable because classic antiresorptive drugs such as bisphosphonates have only minor effects on bone destruction in RA joints.4 Standard anticytokine therapies in RA such as the blockade of TNF-α and IL-6R may also influence bone loss in RA by acting on the RANKL–RANK system.2,4 In this context, the link between pro-inflammatory cytokines and osteoclast formation may explain why anticytokine therapy is effective in inhibiting structural damage in RA.
Periarticular Bone Loss in RA
In addition to bone erosions at the joint margins, patients with RA also may display periarticular bone loss in the trabecular bone in the vicinity of the inflamed joints. The presence of these lesions could indicate that the bone marrow is part of the disease process in arthritis. MRI studies in patients with RA support this notion because they reveal a high prevalence of bone marrow lesions in RA. Histological analysis of such lesions reveals (water-rich) vascularized inflammatory infiltrates, which replace the bone marrow fat and contain aggregates of B and T cells. Of note, similar if not identical MRI changes appear early in the disease process of RA and may predict joint damage in RA.2,7
New Bone Formation in SpA
In RA, there are few signs of repair of bone erosions. At first glance, the paucity of repair is striking because bone formation is usually linked with bone resorption. Thus, increased rates of bone resorption should lead to increased bone formation.7 When compared with RA, the spondyloarthropathies display important differences in the pattern and pathophysiology of joint disease. SpA frequently affects amphiarthroses of the axial skeleton, which lack a true joint cavity and a synovial lining tissue. In addition, entheses, which are sites of tendinous or ligamentous attachments to bone, typically represent the initial sites of inflammation in SpA. In strong contrast to bone erosion in RA, however, bone abnormalities in the SpA are dominated by new bone formation.
Why does bone respond so differently to inflammation in RA and SpA? Although the complex molecular networks have not been fully elucidated, Wnt signaling seems to play a crucial role in determining the effects of arthritis on bone. As described previously, low levels of Wnt activity yield low bone formation and high bone resorption in arthritis, while high levels of Wnt activity can increase bone formation and simultaneously block bone resorption. Indeed, studies indicate that patients with SpA show low levels of the Wnt inibitors Dkk-1 and sclerostin.13,20 Furthermore, Dickkopf-1 blockade can induce fusion of sacroiliac joints in experimental arthritis.21 In contrast, blockade of TNF-α does not appear to inf-luence new bone formation in SpA because TNF-α blockade neither halts new bone formation in experimental animal models of arthritis nor blocks new bone formation in patients with SpA.22 Importantly, TNF-α is a potent down-regulator of bone formation; it is therefore unlikely that blockade of TNF-α reduces new bone formation in SpA. Together, these findings point to a need for new therapeutics to target new bone formation and prevent the ankylosis of joints in SpA.
Bone Marrow Niche
Until now, we have focused on the role of the immune system on bone pathology and the results of bone resorption or new bone formation. In addition to being a target of the immune system in arthritis, however, bone also may promote immunopathogenesis and serve as key player in the orchestra of cells that comprise the immune system.
Among its important direct effects on the immune system, bone provides a special micro-environment, the so-called “bone marrow niche,” a critical site for early B-cell differentiation as well as survival of long-lived B and plasma cells.23 In this context, pre-pro B cells (i.e., the earliest B-cell precursors) and mature plasma cells require CXC chemokine ligand (CXCL)–12 for homing. CXCL-12–expressing cells are a small population of stromal cells that are scattered throughout the bone marrow.24 Of note, these cells allow not only the homing of memory B cells and plasma cells to the marrow but also provide survival signals to increase longevity and promote their activity in establishing and maintaining immunological memory.
Bone seems to be the target and victim from damage inflicted at the hands of the immune system. Indeed, in inflammatory arthritis, bone erosion is the dreaded outcome of the immune system’s destructiveness. As is now realized, however, this view is too simplistic because bone can drive the immune system as well as withstand its blows.
As indicated by a series of intriguing experiments, plasma cells, by means of CXCL 12–induced chemotaxis, can traffic into survival niches in the bone marrow, where they produce antibodies and persist. If bone marrow homing of plasma cells is disturbed, however, a condition observed in murine lupus models where plasma cells are unresponsive to the effects of CXCL-12, a marked accumulation of plasma cells in the spleen, can occur.23 Furthermore, circulating B cells may only become memory B cells if they find appropriate survival conditions outside of the restimulating secondary lymphoid organs. Finally, recent findings on the role of osteoclast-derived cathepsin K in TH17 differentiation and toll-like receptor responses as well as on osteoclast-dependent mobilization of hematopoetic cells from the bone marrow are further evidence for an active role of bone in its duet with the immune system.25,26
Conclusion
In the past, we learned how the immune system can alter the structure and function of bone under physiologic conditions as well as during the course of inflammatory disease. These findings have led to the development of therapies for patients with rheumatic diseases that can reduce deleterious effects in the skeleton, whether bone erosion in the joint or periarticular bone loss and diffuse osteoporosis. We are now learning about the role of the other member of the duet and have gained important insights into how bone affects the immune system. Hopefully, the new knowledge of the roles of both members of the duet will deepen our understanding of the pathogenesis of rheumatic disease and lead to the development of new and more effective approaches to the treatment of the wide range of rheumatic diseases.
Dr. Beyer is an associate faculty member in the department of internal medicine at the University of Erlangen in Nuremberg, Germany. Dr. Schett is a professor at the Institute for Clinical Immunology at the University of Erlangen.
References
- McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429-442.
- Schett G. Osteoimmunology in rheumatic diseases. Arthritis Res Ther. 2009;11:210.
- Schett G, David JP. The multiple faces of autoimmune-mediated bone loss. Nat Rev Endocrinol. 2010;6:698-706.
- Takayanagi H. Osteoimmunology: Shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007;7:292-304.
- Arron JR, Choi Y. Bone versus immune system. Nature. 2000;408:535-536.
- Kong YY, Feige U, Sarosi I, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304-309.
- Schett G, Saag KG, Bijlsma JW. From bone biology to clinical outcome: State of the art and future perspectives. Ann Rheum Dis. 2010;69:1415-1419.
- Lacey DL, Timms E, Tan HL, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165-176.
- Yasuda H, Shima N, Nakagawa N, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95:3597-3602.
- Wong BR, Rho J, Arron J, et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem. 1997;272:25190-25194.
- Zwerina J, Redlich K, Polzer K, et al. TNF-induced structural joint damage is mediated by IL-1. Proc Natl Acad Sci U S A. 2007;104:11742-11747.
- Sato K, Suematsu A, Okamoto K, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673-2682.
- Diarra D, Stolina M, Polzer K, et al. Dickkopf-1 is a master regulator of joint remodeling. Nat Med. 2007;13:156-163.
- Lories RJ, Derese I, Luyten FP. Modulation of bone morphogenetic protein signaling inhibits the onset and progression of ankylosing enthesitis. J Clin Invest. 2005;115:1571-1579.
- Bromley M, Woolley DE. Chondroclasts and osteoclasts at subchondral sites of erosion in the rheumatoid joint. Arthritis Rheum. 1984;27:968-975.
- Gravallese EM, Manning C, Tsay A, et al. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum. 2000;43:250-258.
- Bone HG, Bolognese MA, Yuen CK, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women. J Clin Endocrinol Metab. 2008;93:2149-2157.
- McClung MR, Lewiecki EM, Cohen SB, et al. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med. 2006;354:821-831.
- Cohen SB, Dore RK, Lane NE, et al. Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: A twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheum. 2008;58:1299-1309.
- Appel H, Ruiz-Heiland G, Listing J, et al. Altered skeletal expression of sclerostin and its link to radiographic progression in ankylosing spondylitis. Arthritis Rheum. 2009;60:3257-3262.
- Uderhardt S, Diarra D, Katzenbeisser J, et al. Blockade of Dickkopf (DKK)-1 induces fusion of sacroiliac joints. Ann Rheum Dis. 2010;69:592-597.
- Schett G, Stolina M, Dwyer D, et al. Tumor necrosis factor alpha and RANKL blockade cannot halt bony spur formation in experimental inflammatory arthritis. Arthritis Rheum. 2009;60:2644-2654.
- Tokoyoda K, Egawa T, Sugiyama T, Choi BI, Nagasawa T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity. 2004;20:707-718.
- Erickson LD, Lin LL, Duan B, Morel L, Noelle RJ. A genetic lesion that arrests plasma cell homing to the bone marrow. Proc Natl Acad Sci U S A. 2003;100:12905-12910.
- Asagiri M, Hirai T, Kunigami T, et al. Cathepsin K-dependent toll-like receptor 9 signaling revealed in experimental arthritis. Science. 2008;319:624-627.
- Kollet O, Dar A, Shivtiel S, et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med. 2006;12:657-664.