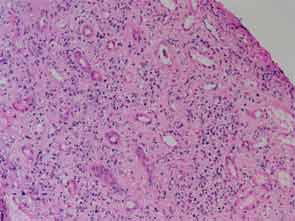
Six months later, the patient developed pruritus and jaundice. His serum bilirubin was 7.9 mg/dL (direct 5.1 mg/dL). Other liver function tests are shown in Table 1. An endoscopic retrograde cholangiopancreatogram showed a mildly dilated distal common bile duct but otherwise normal intra- and extrahepatic biliary systems. A liver biopsy revealed only nonspecific findings: a sparse chronic inflammatory infiltrate confined to the portal spaces and preservation of the bile ducts without significant fibrosis, piecemeal necrosis, or iron overload. The cholestasis was attributed to severe papillary stenosis. His cholestasis resolved following papillary sphincterotomy and the placement of pancreatic stents, which were later removed.

At a follow-up examination two months later, the patient was noted to have diffuse lymphadenopathy, anemia, and an elevated erythrocyte sedimentation rate, raising the possibility of a lymphproliferative disorder. However, a biopsy of an enlarged right axillary lymph node was interpreted as “reactive hyperplasia,” with follicular hyperplasia and reactive plasmacytosis. A bone marrow biopsy showed normal hematopoiesis, and there was no evidence of lymphomatous infiltration, myeloma, or intrinsic marrow pathology.
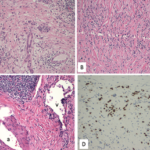
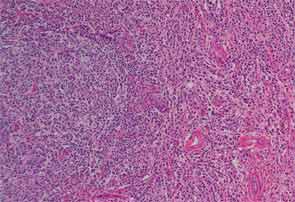
Finally, more than two years after his initial presentation with acute renal dysfunction, recurrent submandibular gland enlargement led to an excisional biopsy. This revealed a Küttner’s tumor (see Figure 3).
Which diagnosis can explain this patient’s multisystem condition?
Answer: Immunoglobulin G4–related disease (IgG4-RD).
The Diagnostic Test
The diagnosis was made following a careful review of the submandibular gland biopsy. Both the histopathology slides and additional immunostains of the existing tissue sample (re-cuts of the tissue block) were essential to establishing the diagnosis.
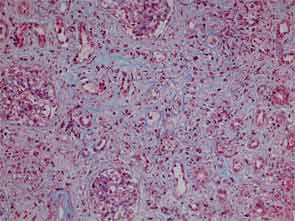
The submandibular biopsy revealed a gland largely replaced by a lymphoplasmacytic infiltrate: reactive lymphoid follicles surrounded by small lymphocytes and plasma cells (see Figure 3A). In addition, there was striking storiform fibrosis (see Figure 3B), obliterative phlebitis, and scattered eosinophils. Immunostaining of the tissue for IgG4 and IgG demonstrated more than 100 IgG4-positive plasma cells per high-power field and an IgG4:total IgG ratio of 0.92. This biopsy was diagnostic of IgG4-RD.
What Is IgG4-RD?
IgG4-RD is a fibroinflammatory condition characterized by a tendency to form tumefactive lesions, a dense lymphoplasmacytic infiltrate rich in IgG4-positive plasma cells, storiform fibrosis, and often, but not always, elevated serum IgG4 concentrations.1 IgG4-RD was not recognized to be an overarching, systemic condition until 2003, when extrapancreatic manifestations were identified in patients with autoimmune pancreatitis.2 The first use of the term “autoimmune pancreatitis” was in 1995.3 Autoimmune pancreatitis, known by a variety of other names up until then, including sclerosing pancreatitis, was linked to elevated serum IgG4 concentrations in 2001.4
Is This a New Disease?
No. Scrutiny and reinterpretation of the medical literature in light of the emerging knowledge of this newly recognized disorder indicates that IgG4-RD has been known by other names, generally while being regarded as an entity isolated to an individual organ system. A disorder termed “multisystemic fibrosclerosis” in the 1960s probably represents—in most cases—IgG4-RD. Table 2 displays a list of previously recognized conditions known by other names that comprise (or may comprise) parts of the IgG4-RD spectrum.

Common Clinical Manifestations
IgG4-RD has now been described in virtually every organ system: the biliary tree, salivary glands, periorbital tissues, kidneys, lungs, lymph nodes, meninges, aorta, breast, prostate, thyroid, pericardium, and skin. A list of the common clinical manifestations in a broad variety of organ systems is shown in Table 3. The histopathologic features of this disease bear striking similarities across organs, regardless of the site of involvement.1 IgG4-RD is therefore analogous to sarcoidosis, another systemic disease in which diverse organ manifestations are linked by unique histopathology.
Patients with IgG4-RD have a predilection for forming mass lesions within organs. Thus, pseudotumors often lead to the patient’s clinical presentation and can lead to misdiagnoses of cancer. These are reported commonly in the orbital region, salivary glands, lung, kidney, lymph nodes, retroperitoneum, and other organs of patients with IgG4-RD.5 Many of these pseudotumors follow an indolent course, but local tissue destruction including erosion of bone has been reported.6 IgG4-RD can also cause more diffuse infiltrative lesions that involve the meninges, skin, or aorta. Aortic aneurysms and dissections can also occur, illustrative of the capacity of IgG4-RD to cause tissue-destructive lesions.5
Another feature of IgG4-RD is its association with allergic or atopic manifestations. Patients with IgG4-RD often have longstanding histories of allergic rhinitis, sinusitis, asthma, and other clinical features of this nature. Many patients have substantial elevations of serum IgE concentrations as well. Peripheral eosinophilia, sometimes on the order of 25% of the total white blood cell count, can be observed. As noted, mild to moderate eosinophil infiltration is also typical of tissue lesions.7
Specific Pathological Features of IgG4-RD
Organ involvement in IgG4-RD is characterized by a diffuse lymphoplasmocytic infiltration and an abundant presence of IgG4-positive plasma cells. Other features are storiform fibrosis, tumefactive lesions, obliterative phlebitis, and, as noted, a modest eosinophil infiltrate.8,9 The inflammatory infiltrate consists of B lymphocytes organized in germinal centers. Ample numbers of T cells are found throughout other parts of the affected tissue. Although IgG4-positive plasma cells are critical to the diagnosis, T cells comprise the dominant cell type in this disease.1
The finding of substantial numbers of IgG4-positive plasma cells in the involved tissue (and a high IgG4:total IgG ratio) is essential for the diagnosis of IgG4-RD. However, IgG4-positive plasma cells can be found in a wide variety of other inflammatory conditions as well and are by no means diagnostic of this condition.
The precise number of IgG4-positive plasma cells per high-power field required to provide strong evidence of the diagnosis of IgG4-RD varies somewhat according to the specific organ. A recent international consensus document provides guidance in this area.10 In salivary glands, for example, a minimum number of more than 100 IgG4-positive plasma cells are histologically highly suggestive of IgG4-RD.
In contrast, in a region such as the retroperitoneum, where the diagnosis of retroperitoneal fibrosis is usually not considered until the process of fibrosis is fairly advanced, there may be relatively few inflammatory cells of any type present. In such cases, the ratio of IgG4- to total IgG-bearing plasma cells may be more useful. In most tissues, a ratio greater than 0.50 strongly suggests the diagnosis of IgG4-RD.
Is Elevated Serum IgG4 Concentration Diagnostic of IgG4-RD?
No, a diverse array of other conditions can be associated with elevations in the serum IgG4 concentration. However, the higher the concentration of IgG4 in the blood, the greater the suspicion for IgG4-RD, particularly if the patient’s clinical manifestations are consistent with this disorder. Our patient’s serum IgG4 concentration was 2,020 mg/dL (normal<121 mg/dL).
As is true with many serologic assays for newly described conditions, tests for serum IgG4 concentrations have been problematic in some circumstances. One recently described problem with serum IgG4 assays relates to the “prozone phenomenon.” The prozone effect can occur in conditions of large analyte excess when assays such as nephelometry, which are dependent upon immune complex formation, are used. The prozone effect can lead to major underestimates of the quantity of the analyte in question.11
Although the prozone effect accounts for some spuriously low, false-negative results, it seems clear that a significant minority of patients—perhaps fewer than 20% of those who have not been treated—have normal serum IgG4 concentrations in the setting of diagnostic histopathology and immunostaining findings in involved organs.
Are Radiologic Examinations Specific for IgG4-RD?
Radiologic findings in IgG4-RD may mimic those of other diseases. The most common example of this is the tendency of IgG4-RD to mimic malignancies in their presentation as pseudotumors. This fact usually makes histopathological confirmation of the diagnosis essential.
Because IgG4-RD was first recognized in the pancreas, the radiology of IgG4-RD is described most thoroughly in that organ. In the proper clinical setting, the finding of a diffusely enlarged, “sausage-shaped” pancreas with peripancreatic stranding is sometimes sufficient for the diagnosis of type 1 autoimmune (IgG4-related) pancreatitis. A bulging contour of the pancreas is seen in most patients. Peripheral inflammation or fibrosis of the pancreas can be detected on computed tomographic (CT) scanning as a peripheral rim or peripancreatic stranding. In contrast to pancreatic cancer, IgG4-related type 1 autoimmune pancreatitis is usually associated with homogeneous contrast enhancement throughout the pancreas. Pancreatic duct stenosis, obstruction, or irregular narrowing may be observed on imaging studies, and temporary stent placement is required for some patients. Calcifications and pseudocyst formation within the pancreas are unusual in IgG4-RD, in contrast to other forms of chronic pancreatitis.12
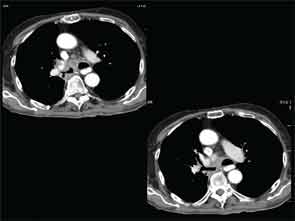
A full understanding of the pulmonary manifestations of IgG4-RD continues to evolve. However, from the standpoint of the CT appearance of lesions, five major types have been described.13 These are nodules, ground-glass opacities, alveolar-interstitial lesions, bronchovascular thickening, and pleural thickening. An example of a pericarinal mass associated with thickening of the bronchovascular bundle and compression of the right mainstem bronchus is shown in Figure 4 (above), along with its corresponding histopathology and immunostaining features.
The diversity of pulmonary lesions in IgG4-RD broadens significantly the differential diagnosis that the rheumatologist must consider when evaluating a patient with lung problems. Nodular lesions may represent lung tumor, an opportunistic infection, granulomatosis with polyangiitis (Wegener’s granulomatosis), and other entities—but also IgG4-related lung disease. Similarly, ground-glass opacities may indicate fluid overload, viral infection, alveolar hemorrhage, bronchoalveolar carcinoma, and other diagnoses, including IgG4-related lung disease. Even honeycombing, part of the advanced stage of interstitial lung disease and nonspecific or usual interstitial pneumonitis that characterizes a broad swath of rheumatologic disease that includes rheumatoid arthritis, systemic sclerosis, inflammatory myopathies, and antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis occurring in the setting of antimyeloperoxidase ANCA, may also be the result of IgG4-RD. Finally, confronted with the striking thickening of the bronchovascular bundle that can be caused by IgG4-RD, the rheumatologist must also consider lymphomatoid granulomatosis, multicentric Castleman’s disease, and sarcoidosis, among others.
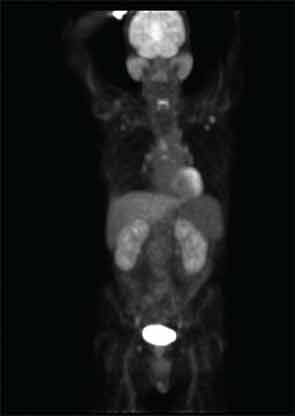
Positron emission tomography (PET) with CT can be useful to determine the extent of disease and may also be helpful to gauge disease activity and response to treatment. Our patient underwent a PET/CT that revealed unanticipated findings in the lung, demonstrating bronchiectasis, ground glass opacities, and pulmonary nodules (see Figure 5, at right) in addition to showing increased tracer uptake in his parotid glands and lymph nodes.
What Is the Treatment for IgG4-RD?
Glucocorticoids are the first-line treatment for IgG4-RD. Many cases require aggressive and immediate treatment to prevent organ dysfunction and failure. However, other disease manifestations (e.g., IgG4-related lymphadenopathy) may remain indolent and relatively asymptomatic for years.14 Therefore, the treatment regimens must be individualized for each patient. One approach to the use of glucocorticoids is to treat patients initially with prednisolone at a dose of 0.6 mg per kilogram of body weight per day for two to four weeks and then to taper the steroids over a period of three to six months.15 Some investigators, wary of the long-term complications of glucocorticoids, have aimed to discontinue these agents after three months.16 Agents such as azathioprine, mycophenolate mofetil, and methotrexate have been employed as potential glucocorticoid-sparing agents or remission–maintenance drugs, but their true efficacy in these roles is unclear.
For patients with IgG4-RD that is refractory to glucocorticoids, patients that are unable to taper below a desired dose, or patients with recurrent IgG4-RD, B-cell depletion with rituximab may represent a promising approach to treatment.17 Patients treated with rituximab have generally demonstrated prompt clinical and serological responses, with the ability to taper glucocorticoids rapidly and a swift decline in serum IgG4 concentrations. Moreover, the decline in serum IgG4 concentrations appears to be isolated, targeting this IgG subclass alone while leaving the concentrations of IgG1, IgG2, and IgG3 stable.18 This finding suggests that one potential mechanism of rituximab’s efficacy in IgG4-RD is through interference with the repletion of short-lived plasma cells that are producing IgG4 in this condition. Additional studies of this hypothesis are required, however, and an ongoing clinical trial will investigate these concepts further.
Summary
Our patient had a multiorgan system disease of at least two years’ duration characterized by tubulointerstitial nephritis, salivary gland enlargement, jaundice, diffuse lymphadenopathy, and an elevated erythrocyte sedimentation rate. In addition, it is likely that his pancreatitis, which led to glucose intolerance, was in fact IgG4-related (type 1) autoimmune pancreatitis. He was misdiagnosed with a number of other conditions before the correct diagnosis of IgG4-RD was recognized.
IgG4-RD is a newly recognized entity, but in fact it is an old one. The disease can involve multiple organ systems, frequently in a metachronous fashion (i.e., first one organ, then another, and then another). The disease tends to cause mass-forming lesions that can mimic cancer, infections, and rheumatic conditions such as granulomatosis with polyangiitis (Wegener’s) and SS. This disease is responsible for substantial percentages of cases—and sometimes all cases—of a variety of clinical entities previously referred to by other names (e.g., retroperitoneal fibrosis, Riedel’s thyroiditis, Mikulicz disease, Küttner’s tumor, and others).
Most patients respond well to glucocorticoids, but many are unable to discontinue these medications without disease recurrence. B-cell depletion is a promising treatment strategy now under investigation.
Follow-up
Because of his glucose intolerance and the extensive nature of his disease, we treated our patient with rituximab 1 gram intravenously given on two separate occasions. Within one month of his first dose, his parotid gland swelling had resolved, and his serum IgG4 concentration had declined by more than 600 mg/dL. He is currently enrolled in an ongoing clinical trial.
Dr. Atac is a visiting researcher at the Massachusetts General Hospital. Dr. Stone is a professor of medicine at Harvard Medical School and director of clinical rheumatology at Massachusetts General Hospital in Boston.
References
- Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366:539-551.
- Kamisawa T, Egawa N, Nakajima H. Autoimmune pancreatitis is a systemic autoimmune disease. Am J Gastroenterol. 2003;98:2811-2812.
- Yoshida K, Toki F, Takeuchi T, et al. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci. 1995;40:1561-1568.
- Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344:732-738.
- Khosroshahi A, Stone JH. A clinical overview of IgG4-related systemic disease. Curr Opin Rheumatol. 2011;23:57-66.
- Pace C, Ward S. A rare case of IgG4-related sclerosing disease of the maxillary sinus associated with bone destruction. J Oral Maxillofac Surg. 2010;68:2591-2593.
- Kamisawa T, Anjiki H, Egawa N, Kubota N. Allergic manifestations in autoimmune pancreatitis. Eur J Gastroenterol Hepatol. 2009;21:1136-1139.
- Zen Y, Nakanuma Y. IgG4-related disease: A cross-sectional study of 114 cases. Am J Surg Pathol. 2010;34:1812-1819.
- Deshpande V, Gupta R, Sainani N, et al. Subclassification of autoimmune pancreatitis: A histologic classification with clinical significance. Am J Surg Pathol. 2011;35:26-35.
- Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012; 25:1181-1192.
- Khosroshahi A, Cheryk LA, Carruthers MN, et al. Prozone phenomenon leads to low IgG4 concentrations in IgG4-related disease (Abstract 2527). Arthritis Rheum. 2012;64(10 Suppl):S1067.
- Chang WI, Kim BJ, Lee JK, et al. The clinical and radiological characteristics of mass-forming autoimmune pancreatitis: Comparion with chronic pancreatitis and pancreatic cancer. Pancreas. 2009;38:401-408.
- Zen Y, Inoue D, Kitao A et al. IgG4-related lung and pleural disease: A clinicopathologic study of 21 cases. Am J Surg Pathol. 2009;33:1886-1893.
- Cheuk W, Yuen HK, Chu SY, et al. Lymphadenopathy of IgG4-related sclerosing disease. Am J Surg Pathol. 2008;32:671-681.
- Kamisawa T, Shimosegawa T, Okazaki K, et al. Standard steroid treatment for autoimmune pancreatitis. Gut. 2009;58:1504-1507.
- Ghazale A, Chari ST, Zhang L, et al. Immunoglobulin G4-associated cholangitis: Clinical profile and response to therapy. Gastroenterology. 2008;134:706-715.
- Khosroshahi A, Carruthers MN, Deshpande V, et al. Rituximab for the treatment of IgG4-related disease: Lessons from 10 consecutive patients. Medicine (Baltimore). 2012;91:57-66.
- Khosroshahi A, Bloch DB, Deshpande V, Stone JH. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 2010;62:1755-1756.
- Stone JH, Khosroshahi A, Deshpande V, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum. 2012;64:3061-3067.