Shidlovski/shutterstock.com
CHICAGO—Caryn A. Libbey, MD, clinical associate professor of medicine at Boston University School of Medicine, described the evolving in our understanding of amyloid at the ACR’s State-of-the-Art Clinical Symposium in April. Amyloidosis is a rare disease that is often underdiagnosed and undertreated.
“Even though this disease has been around for 150 years, I still consider it a relatively modern disease, because the first fibril was identified only in 1971,” explained Dr. Libbey. There are more than 30 amyloid precursor proteins, which, in healthy individuals, remain soluble. When proteins, such as the kappa or lambda light chain or transthyretin, become insoluble, they can cause amyloidosis.
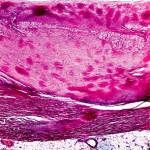
The insoluble molecules can be visualized in the tissue as nonbranching beta pleated sheets that stain positive for Congo red. Unless the stain is performed properly, there may be a 10–15% false-positive rate and a 10–15% false-negative rate. Unfortunately, the stain is infrequently performed because pathologists do not often have amyloidosis as a differential diagnosis unless the clinician has alerted the pathologist. Diagnosis of amyloid is made only when a biopsy is positive.
Types of Amyloid
Local amyloid—This type may form without systemic deposition in skin, the larynx/bronchus or bladder, or less often, the breast or lung.
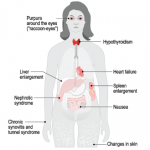
AL (amyloid from light chain)—AL patients frequently experience malaise, weight loss and fatigue. They often have proteinuria with or without decreased glomerular filtration rate. Approximately 45% of patients with amyloidosis have cardiac symptoms that present as congestive heart failure, restrictive cardiomyopathy, edema, arrhythmia and/or heart block. In addition, approximately one-third of patients have neuropathy that presents in peripheral sensory and motor nerves or in autonomic nerves. Foot pain is especially common in these individuals, for which a patient may be referred to rheumatology.
AA (amyloid from serum amyloid A deposition)—Amyloid A (AA) amyloidosis is more likely to be seen by rheumatologists outside the U.S. It is caused by chronic uncontrolled inflammation and can result from tuberculosis, osteomyelitis, rheumatoid arthritis, ankylosing spondylitis, Crohn’s disease, juvenile idiopathic arthritis and psoriatic arthritis, among other causes (see sidebar below).
ATTRm (amyloid from point mutation of transthyretin)—Most rheumatologists won’t encounter transthyretin-related neuropathic amyloidoses (ATTRm), rare, familial forms of amyloidosis first described in 1952. Most often, ATTRm presents with neuropathy, cardiomyopathy and nephropathy.
More than 120 point mutations have been described that result in familial ATTR, but some mutations are more common than others. A Portuguese mutation has a high penetrance, and thus, most individuals who inherit the gene will express symptoms of disease. The age of onset of individuals with the Portuguese mutation is 25–30 years. These individuals experience progressive peripheral sensorimotor neuropathy, followed by cardiac and renal symptoms. The patient often dies within 10 years of diagnosis, if untreated.
ATTRwt (amyloid from wild type or natural, without mutation of, TTR)—ATTRwt is the most common, but under-recognized form of amyloid, commonly causing new onset of congestive heart failure (CHF) in men older than 75. It’s estimated that 25–30% of the normal population has amyloid cardiac deposition. Of interest from the rheumatology point of view, approximately 50% of ATTRwt patients will have carpal tunnel syndrome (CTS) symptoms 5–10 years before the onset of CHF, so we recommend thinking about ATTRwt when a rheumatologist sees an older male with new onset of CTS.
ALect2 (amyloid from leucocyte chemotactic factor 2)—Researchers discovered leucocyte chemotactic factor 2 (ALect2) amyloidosis in 2008. It is found in individuals of Mexican/Hispanic heritage, and in the U.S. it is thus most frequently diagnosed in New Mexico, Arizona and West Texas. It can also be found in Egypt, Punjab and the First Nations people of Canada. It is diagnosed between the ages of 43 and 88 years, and patients present with slowly progressive renal failure, which, after approximately five years, leads to end-stage renal disease. These patients typically do not have proteinuria.
Diagnosing Amyloidosis
In many cases, patients with amyloidosis have a normal result on serum protein electrophoresis (SPEP) test, so this test is not particularly useful for diagnosis. In addition to serum and urine immunofixation, the most sensitive test is the free light chain assay, and Dr. Libbey recommends these tests for diagnosis when looking for plasma cell dyscrasia associated with AL amyloid.
Patients with cardiac involvement may have an electrocardiogram that reveals arrhythmia and low voltage, and shows thickened septal measurements. These patients could be imaged with a cardiac MRI with late gadolinium enhancement, as well as a technetium TC 99M pyrophosphate Tc99-pyrophosphate scan. This scan may distinguish AL from ATTR cardiomyopathy.
If a physician diagnoses amyloidosis with cardiac involvement, the patient should not be prescribed digoxin or calcium channel blockers; these medications bind to fibril and potentiate toxicity. Likewise, if a patient with mild congestive heart failure is responding poorly to these drugs, it may be because of the presence of amyloid in the heart.
The only way to make a diagnosis of amyloid is with a tissue biopsy. It is recommended to start with an abdominal fat cell aspirate, which can be done at the bedside.
Dr. Libbey concluded her presentation by explaining that effective treatments are now available for most kinds of amyloid, if it is diagnosed early in the course of the disease. AL can be treated with autologous stem cell transplant or chemotherapy aimed at the plasma cell dyscrasia. AA can be treated with aggressive management of the underlying disease, ATTR can be slowed by a TTR stabilizer, such as diflunisal or tafamidis in Europe, or patients may be considered for liver transplant. A variety of novel treatments are being studied, which will change the future of this not-so-rare disease.
Nevertheless, amyloidosis “is still a missed diagnosis.” Dr. Libbey encouraged rheumatologists to add it to their list of differential diagnoses.
Lara C. Pullen, PhD, is a medical writer based in the Chicago area.
Causes of AA Amyloid
- Inflammatory: rheumatoid arthritis, ankylosing spondylitis, Crohn’s disease, juvenile idiopathic arthritis, psoriatic arthritis, colitic arthropathy
- Infectious: tuberculosis, osteomyelitis, leprosy, substance users/skin poppers, bronchiectasis
- Autoinflammatory: familial Mediterranean fever, tumor necrosis factor receptor-associated periodic syndrome, cryopyrin-associated periodic syndrome, hyperimmunoglobulinemia D with periodic fever syndrome
- Tumor: renal cell carcinoma, atrial myxoma, Hodgkins, IgG4 disease