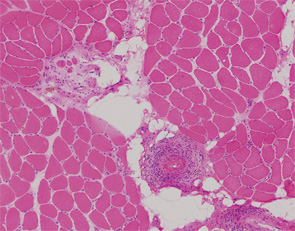
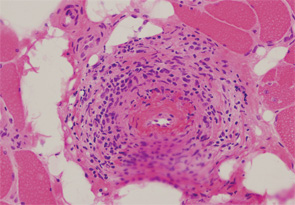
Patients with diffuse systemic sclerosis (dSS) with an associated ANCA-positive vasculitis have been infrequently described. In this article, we report the case of a 53-year-old woman, previously diagnosed with dSS, who developed an ANCA-associated vasculitis, which manifest as peripheral nerve and muscle disease.
Scleroderma is a disease characterized by sclerosis of the skin and visceral organs, vasculopathy, Raynaud’s phenomenon and the presence of autoantibodies. The disease spectrum is wide, with systemic and localized forms, depending on the extent of skin thickening. There is also a vasculopathy involving multiple organ systems that leads to the presence of cutaneous and mucosal telangiectasias, digital ulcers and tissue ischemia. Medium-size vessels can also be involved, sometimes leading to scleroderma renal crisis or pulmonary artery hypertension. However, scleroderma is rarely associated with an ANCA-positive vasculitis. We report this unique situation.



