IL-6 levels were markedly elevated in patients with ECD who underwent analysis of their cytokine profile.5 It was further noted that patients treated with IFN-α normalized their serum levels of IL-6.5 Tocilizumab is a humanized monoclonal antibody that inhibits the IL-6 receptor. Therefore, anti-IL-6 therapy with tocilizumab (TCZ) was used to treat two cases of multisystem ECD.
In our cases, we show dramatic improvement with anti-interleukin-6 therapy and think it warrants more study. Current therapy is suboptimal, & there is a role for alternative treatments in ECD.
Case Series
Two patients referred to the rheumatology service met criteria for diagnosis of Erdheim-Chester based on a combination of clinical and radiographic features. Findings from standard clinical, radiologic and histopathologic techniques are described.
(click for larger image)
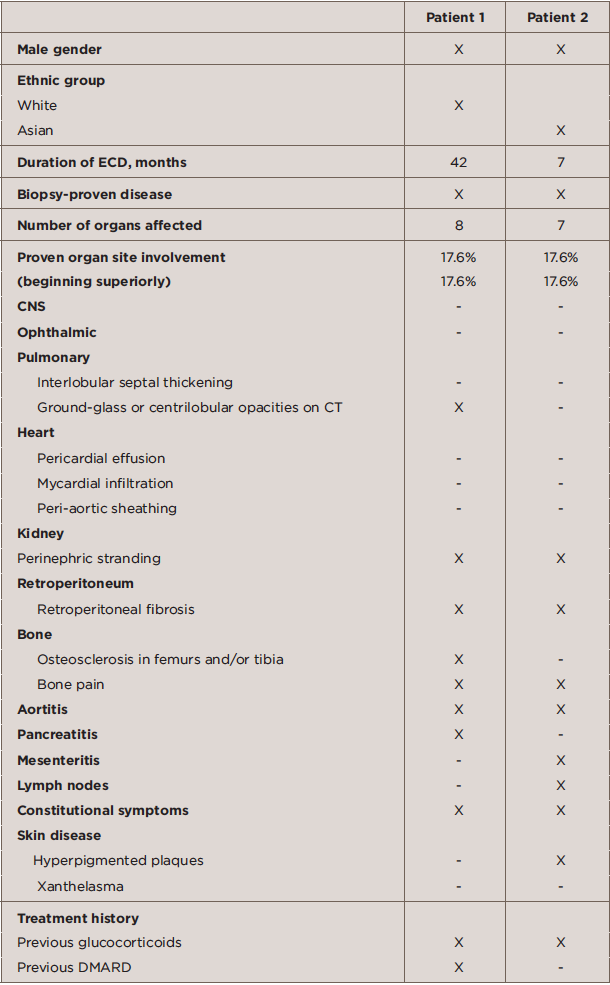
TABLE 1: Baseline Characteristics of ECD Patients
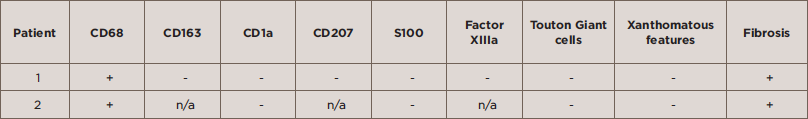
The clinical history of Patients 1 and 2 is described below. The baseline characteristics of Patients 1 and 2 are described in Table 1, and the immunohistochemical features of the two patients are shown in Table 2.
Patient 1: A 59-year-old male initially presented to an emergency department with right wrist drop, and numbness and tingling of his fingers. He had a medical history of hypertension and type 2 diabetes mellitus. He had a prior history of tonsillectomy. Medications included oral hypoglycemics and furosemide. Family history included myelodysplastic syndrome in his mother and coronary artery disease in his father. He worked as an accountant and was married with two children. He smoked one pack per day and had moderate alcohol consumption. Review of symptoms included an unintentional 8 kg weight loss, fatigue, chills, mild dyspnea on exertion, nausea and anorexia.
His vital signs were normal. Physical exam was notable for tanned appearance, hepatosplenomegaly, mild lower extremity edema and right wrist drop with strength of 3 out of 5 during dorsiflexion. Sensory exam was normal, as well as the remaining motor exam.
Laboratory studies revealed hematocrit of 25%, platelet count was 425 x 109 cells per liter (normal <350) and creatinine of 265 μmol/L (from a baseline creatinine of 106 μmol/L). Liver enzymes were normal.
ESR was 107 mm/hour (normal range 1–20). Urinalysis showed no protein, RBCs and WBCs, no granular casts. Peripheral flow cytometry revealed lymphopenia. Serum protein electrophoresis was normal. Tests for antinuclear antibody and anti-neutrophil cytoplasmic antibody were negative. Immunoglobulin G (IgG) subclasses were normal with IgG4 level of 0.390 g/L (normal
Brain MRI was normal, and the patient’s wrist weakness improved throughout his hospital admission. The weight loss, new renal insufficiency and anemia were concerning, and led to further imaging to look for occult malignancy. A CT scan of chest, abdomen and pelvis were abnormal: the main concerns were a T10 sclerotic lesion, multifocal ground-glass opacities of the lungs, as well as diffuse pancreatic enlargement with an intra-abdominal fluid collection (see Figures 1A and B).
Biopsy of the bone was non-diagnostic, with adequate samples of the pancreas and lung (see Figures 2A and B). Bone marrow biopsy was normal. PET-CT revealed uptake in lung ground-glass opacities, proximal humeri, femoral heads, greater trochanter and head, and body and tail of the pancreas (see Figure 2C).
Intermittent prednisone pulses were initiated by a local oncologist to treat his systemic inflammatory condition. Azathioprine was prescribed between July 2011 and December 2011, until the patient was hospitalized for Legionella pneumonia. He subsequently developed transaminitis, and a liver biopsy revealed nonspecific changes. The lung disease did not resolve, and he was referred to a pulmonologist.
(click for larger image)
TABLE 2: Histopathologic Features of Patients with Erdheim-Chester
Because of concern for IgG4-related disease, the lung biopsy tissue was retrospectively stained for IgG4+ plasma cells. Referral to the MGH Division of Rheumatology was made. The IgG4+ cell count was 21/hpf with 43% IgG4/IgG ratio. All available pathologic material, including specimens from the pancreas, liver, lung and bone marrow, was reviewed by Vikram Deshpande, MD.
The lung biopsy showed CD68-positive, S-100-negative, CD1a-negative foamy histiocytes not previously appreciated. All were consistent with a diagnosis of ECD.
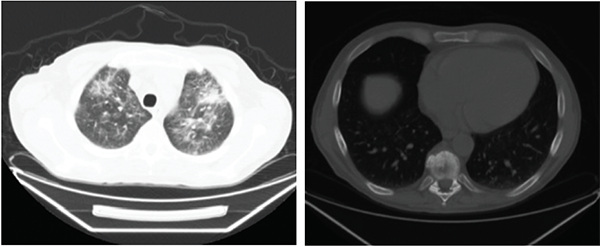
Figures 1A (left) & B (right): Patient 1
A clinical decision was made to start 8 mg/kg tocilizumab intravenously every four weeks; prednisone was tapered from 20 mg to 10 mg daily over a three-month period. A repeat PET-CT showed lungs with markedly less uptake and stable fludeoxyglucose avidity in the pancreas and bone (see Figure 2D). Repeat CT scans showed vast improvement in inflammation intra-abdominally and complete resolution of pulmonary ground-glass opacities (see Figure 4A).

Figures 1C (left) & D (right): Patient 2
Figures 1A–D show the computed tomography (CT) on Patients 1 and 2. 1A is a CT scan of Patient 1 without contrast showing pulmonary ground-glass opacities. 1B is a CT scan of Patient 1 showing a sclerotic lesion at T10. 1C is a CT scan of Patient 2 with contrast showing soft-tissue rind around the kidneys, also seen in Patient 1. 1D is a CT scan of Patient 2 with contrast showing soft-tissue infiltration around the abdominal aorta.
The patient has been in remission without repeat hospitalizations for five years.
Patient 2: A 54-year-old man presented in 2014 to his physician with back and flank pain, low-grade fever and a 7 kg weight loss. Medical history included resolved depression treated with antidepressants, no prior surgeries and no allergies. His family history was unremarkable. He was a married veterinarian with two children and a non-smoker with occasional alcohol use.
On review of systems, he had low-grade fevers, back pain, flank pain and pain in the rhomboid area. He had no neurological complaints, vision symptoms, swollen lymph nodes or cardiopulmonary symptoms. He denied urinary hesitancy or other voiding symptoms. There were no rashes.
Vital signs were normal. Positive findings included lacrimal gland enlargement, goiter (left greater than right side) and several enlarged lymph nodes (up to 2 cm in his axilla and 1.5 cm in the inguinal area). His abdomen was mildly distended, nontender, with no organomegaly. There was no costovertebral angle tenderness. There were hyperpigmented plaques on his distal extremities. Laboratory studies revealed an increased creatinine level, and an ultrasound demonstrated bilateral hydronephrosis. Bloodwork in September 2015 showed WBC 10.7 x 109/L, Hct 35%, platelets 348 x 109 (normal <350), creatinine 98 μmol/L, total protein 60 g/L, albumin 33 g/L, ALP 164 U/L, AST 35 U/L, ALT 74 U/L and high-sensitivity C-reactive protein (HS-CRP) 105 mg/L (normal <5). A lipid panel was normal. He was hepatitis B immune. Urinalysis showed no protein, trace hemoglobin, 11–30 WBCs per HPF, 1–3 RBCs per HPF and a few epithelial cells. The patient’s IgG subclasses were normal.
Abdominal MRI showed bilateral hydronephrosis, with abnormalities in the vicinity of the renal pelvis, renal sinuses, and proximal ureters suggestive of either extensive infiltrating carcinoma or retroperitoneal fibrosis (there was abdominal aortic soft-tissue encasement along with renal pelvis, ureter involvement, and rind around the kidneys). Initial rheumatologic evaluation was concerning for ECD; however, differential diagnoses included histiocytosis, amyloidosis, IgG4-RD or idiopathic retroperitoneal fibrosis.

Figures 2A (left) and B (right)
He was hospitalized, and an inpatient CT abdomen showed abnormal soft-tissue attenuation surrounding the infrarenal aorta, inferior vena cava, renal vessels, and ureters causing severe bilateral hydronephrosis and severe bilateral perirenal stranding (see Figures 1C and D, below). There was also an abnormal area of soft-tissue attenuation in the central mesentery. A nonspecific 12 mm lytic lesion in the L2 vertebral body was noted. He underwent placement of nephrostomy tubes and at the same time had laparoscopic biopsy of retroperitoneal tissue. Pathology showed an inflammatory lesion with a lymphohistiocytic infiltrate including foamy histiocytes (see Figure 3A). Immunohistochemical staining showed these histiocytes were positive for CD68 and BRAF and negative for CD1a and S100, most consistent with a diagnosis of ECD (see Figures 3B and C, p. 22). Only rare plasma cells expressed IgG4 (see Figure 3D).
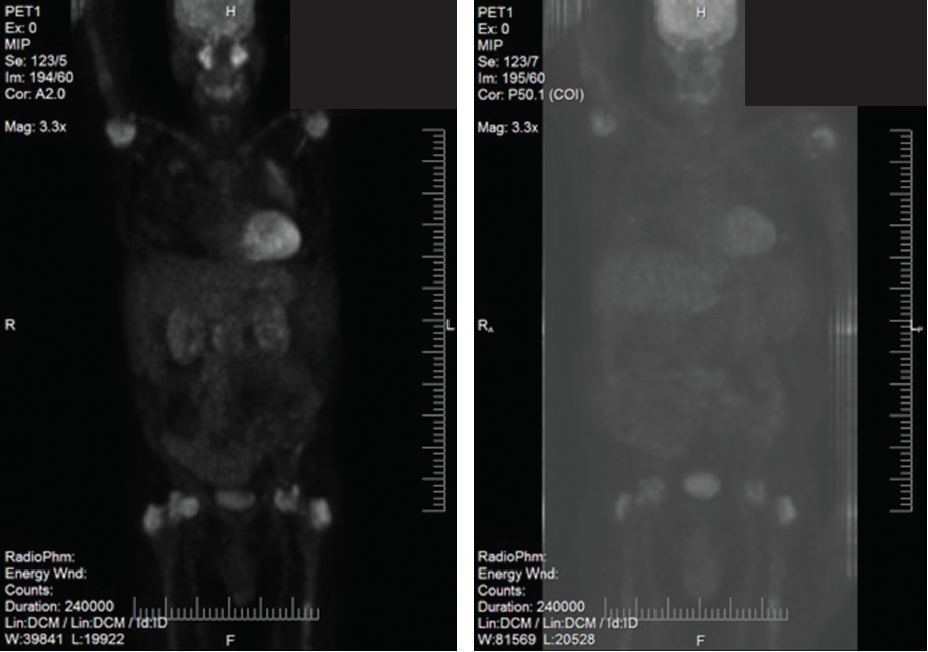
Figures 2C (left) and D (right)
Figures 2A–D show the histopathologic findings and PET-CT findings for Patient 1. 2A and 2B are low- and high-power views (A and B, respectively) of hematoxylin and eosin (H&E) stained lung sections, demonstrating inflammatory infiltration, which includes foamy histiocytes. 2C is a PET-CT prior to treatment with tocilizumab and shows uptake in the lungs, symmetric uptake in proximal humeri, femoral heads and greater trochanter, as well as activity in the head, body and tail of the pancreas. 2D is a PET-CT done at four months of treatment with tocilizumab and shows the lungs with less uptake; no FDG avidity in the pancreas, and stable bone uptake.
TCZ 8 mg/kg IV every four weeks was initiated. His course was complicated by recurrent urinary tract infections. There has been complete resolution of flank pain. A CT scan in January 2016 showed no progression of retroperitoneal soft tissue attenuation around the kidneys, renal veins and aorta. The prominent stranding in the mesentery has decreased compared with his prior CT scan (see Figures 4B and C).
ECD has a male predominance & is typically diagnosed in adults between ages 40 & 70. Diagnosis is often delayed due to its rarity, multiple disease presentations & lack of awareness.
Discussion
We conducted a systematic literature review to expand on the discussion of these two case presentations. Note: We searched the following databases: Scopus, PubMed and Cochrane Library. Our literature search combined the terms Erdheim-Chester disease with treatment. An additional search strategy included Erdheim-Chester disease with tocilizumab.
ECD is a rare, potentially life-threatening condition, which recently has been better understood.2,6 Recent consensus guidelines have helped in both the diagnosis and management of this disease.2 Multiple therapeutic approaches have been used for ECD, but treatment is limited to Grade C and D evidence based on a lack of randomized controlled trials and few limited prospective therapeutic studies.2 Historically, such therapies as vinca alkaloids, anthracyclines, cyclophosphamide and high-dose chemotherapy with autologous stem cell transplantation, were utilized in small series with poor outcomes.2 Corticosteroids were found to be ineffective as monotherapy or in inducing remission; however, clinical experience supports their use to decrease acute inflammation.2
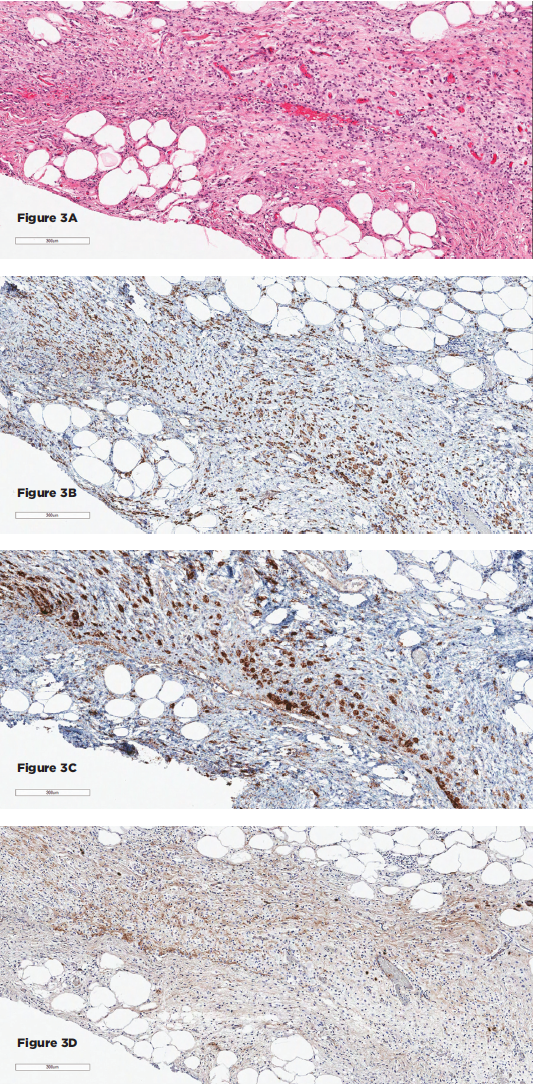
(click for larger image)
Figures 3A–D show the histopathologic findings for Patient 2. 3A–D show sections of the retroperitoneal lesion showing a fibrohistocytic lesion with foamy histiocytes on H&E (3A), positive for CD68 (3B) and BRAF (3C), negative for S100 and CD1a (not shown), and with only rare IgG4+ plasma cells (3D).
First-line therapy remains interferon therapy (IFN-α), based on multiple case series showing a survival benefit.2 However, interferon treatment has major limitations, which include the lack of efficacy, particularly in the presence of cardiovascular and CNS involvement, as well as an unfavorable side effect profile.7,8 Second-line therapies have included cladribine, sirolimus, infliximab, anakinra, tyrosine kinase inhibitors and vemurafenib.
Sirolimus (SRL) is an inhibitor of the mammalian target of rapamycin (mTOR) and has been used in treating malignancies and transplant rejection.3 An observation that a small number of ECD patients who have activating PIK3CA mutations resulting in mTOR pathway activation has led to sirolimus as a consideration in treatment of ECD.2,3,9
A recent open-label prospective trial by Gianfreda et al using sirolimus and prednisone was additionally based on the rationale of interference with the immune dysregulation seen in ECD.3 The study enrolled 10 patients. Eight had objective response or disease stabilization, and two had disease progression.3 The reported overall mortality was 20%.3
The number of cases reported using anti-tumor necrosis factor (TNF) therapies is limited. Dagna et al from Italy describe a 56-year-old man with a two-year history of recurrent pericardial effusion and acute cardiac tamponade who was successfully treated with infliximab 5 mg/kg every six weeks.10 Methotrexate was added to reduce the risk of possible autoantibodies.10
A second patient, a 60-year-old man with cardiac involvement, failed IFN-α because of side effects but had a good response to 5 mg/kg infliximab every six weeks.10
This group went on to describe a 54-year-old man with ECD who developed a large pericardial effusion.11 He had failed treatment with prednisone, IFN-α and methotrexate (20 mg/wk). Analysis of the exudative effusion demonstrated foamy histiocytes and increased levels of TNFα, CCL2 and IL-6. The intent was to treat with anti-TNFα (infliximab).11
Anakinra, an IL-1 receptor antagonist (IL-1RA), has been reported in a small case series as a treatment for ECD. Aouba et al describe two patients with ECD treated with anakinra.12 The first patient was a 56-year-old female with fever, fatigue, lumbar pain and eyelid xanthelasma, who failed corticosteroids and cladribine. Symptoms resolved on Day 3 with treatment of 100 mg sc/daily anakinra.
Also, a 55-year-old man with similar symptoms responded on Day 5 of anakinra therapy. Interestingly, CRP and IL-6 levels measured at Month 3 dropped significantly. The IL-1β level increased as expected with an IL-1R blocker, but the TNFα levels at Month 3 were unchanged.12 ECD with cardiac involvement has now been treated with anakinra alone, and a case with CNS involvement was treated with anakinra in combination with glucocorticoids.13,14 In the pediatric population, anakinra was used as an effective treatment modality and was subsequently changed to anti-IL-1β therapy with canakinumab every 10 weeks to decrease the frequency of discomfort associated with daily injections.15
BRAF V600 mutations have been reported in 50–100% of patients with ECD, depending on both the series and methods used.16 Vemurafenib, a small-molecule inhibitor of BRAF harboring V600E or, to a lesser extent, V600K mutations, was used in an open series of eight patients with ECD at a dose of 480–960 mg po BID for up to six months. All patients demonstrated a significant reduction of FDG activity on PET.16 Significant side effects included keratosis pilaris, xerosis, photosensitivity and arthralgias. Treatment outcomes are still limited to a handful of cases, and there is no equivalent therapy for patients with wild-type BRAF. Adverse events: Three of the eight patients developed skin cancers, with one patient developing an infiltrative cutaneous squamous cell carcinoma at Month 5.16
Houston et al initially treated a 69-year-old male with biopsy-proven ECD with anakinra to avoid the side effects of IFN, which led to a reduction in pain within two weeks. At that point, molecular studies confirmed BRAF mutation, and anakinra was discontinued in favor of vemurafenib. The patient had return of constitutional symptoms on vemurafenib alone, so anakinra was restarted. The patient’s symptoms resolved rapidly on this combination, which points to a role for combination therapy in the treatment of ECD.17
Back to Our Cases
Our two cases of ECD were treated with TCZ as described. Our initial case was treated empirically with TCZ and prednisone based on the assumption that ECD manifestations are largely driven by Th-1 mediated immune response. Further, elevated IL-6 levels have been described in blood and tissues in patients with ECD. Both of our patients had widespread disease with manifestations that were similar to those reported in the literature.27
In Patient 1, we noted improvement in symptoms of ECD, as well as clear radiological improvement over a four-month course of using 8 mg/kg tocilizumab IV every four weeks.
In Patient 2, we noted clinical stabilization and some early radiographic improvement.
Known risks of TCZ include immunosuppression, elevated LFTs, cytopenias, elevated cholesterol and, rarely, bowel perforation.18 The most notable side effect in our patients was recurrent urinary tract infections in our second case, which may have been related to the medication or the underlying disease process.
Our clinical results are limited by the small sample size of two patients. The two patients described did not have CNS involvement, which is an independent predictor of death. Also, we did not obtain cardiac imaging in our patients to screen for preclinical cardiac involvement. Advances in cardiac imaging suggest that cardiac involvement may be more common than once thought in ECD, and experts now recommend screening for occult cardiac involvement.16,19
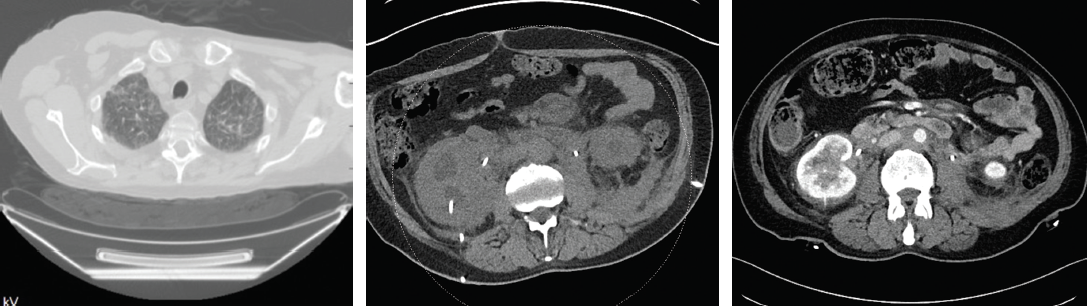
Figures 4A–C demonstrate the response to treatment in Patients 1 and 2. 4A is the CT scan of Patient 1 and demonstrates resolution of the ground-glass opacities in the lungs after treatment. 4B and 4C are the abdominal CT scans of Patient 2 before (4B) and after (4C) treatment.
What Lies Ahead
A prospective, open-label, single-arm, Phase 2, pilot study of tocilizumab in patients with ECD is ongoing. Recruitment began in 2012; unfortunately, recruitment into meaningful prospective trials is made difficult by the rarity of the disease.
In our cases, we show dramatic improvement with anti-IL-6 therapy and think it warrants more study. Current therapy is suboptimal, and there is a role for alternative treatments in ECD.
Stefanie D. Wade, MD, is completing her internal medicine residency at the University of Connecticut Health Center and plans to pursue a fellowship in rheumatology.
Michael A. Seidman, MD, is a pathologist and researcher at St. Paul’s Hospital in downtown Vancouver, Canada.
Edward C. Jones, MD, is a consultant anatomical pathologist at the Vancouver General Hospital and a clinical professor at the University of British Columbia.
Arnold Radu, MD, completed his radiology residency at McGill University and has finished a musculoskeletal fellowship at Vancouver General Hospital.
Ryan Paterson, MD, is an assistant professor in the Department of Urologic Sciences at Vancouver Hospital and Providence Health Care. He graduated UBC for medical school and was an Endourology and Laparoscopy, American Foundation for Urologic Disease Scholar.
Vikram Deshpande, MD, is a pathologist at Massachusetts General Hospital and associate professor at Harvard Medical School in Boston.
John H. Stone, MD, is the chief of Rheumatology at Massachusetts General Hospital and professor at Harvard Medical School in Boston.
Mollie N. Carruthers, MD, completed her rheumatology training at Massachusetts General Hospital and is now a clinical associate professor at the University of British Columbia.
References
- Mazor RD, Manevich-Mazor M, Shoenfeld Y. Erdheim-Chester disease: A comprehensive review of the literature. Orphanet J Rare Dis. 2013 Sep 8;8:137.
- Diamone EL, Dagna L, Hyman DM, et al. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester Disease. Blood. 2014 Jul 24;124(4):483–492.
- Gianfreda D, Nicastro M, Galetti M, et al. Sirolimus plus prednisone for Erdheim-Chester disease: An open label trial. Blood. 2015 Sep 3;126(10):1163–1171.
- Cangi, MG, Biavasco R, Cavalli G, et al. BRAFV600E-mutation is invariably present and associated to oncogene-induced senescence in Erdheim-Chester disease. Ann Rheum Dis. 2015 Aug;74(8):1596–1602.
- Arnaud L, Gorochov G, Charlotte F, et al. Systemic perturbation of cytokine and chemokine networks in Erdheim-Chester disease: A single-center series of 37 patients. Blood. 2011 Mar 10;117(10):2783–2790.
- Haroche J, Arnaud L, Amoura Z. Erdheim-Chester disease. Curr Opin Rheumatol. 2012 Jan;24(1):53–59.
- Arnaud L, Hervier B, Néel A, et al. CNS involvement and treatment with interferon-α are independent prognostic factors in Erdheim-Chester disease: A multicenter survival analysis of 53 patients. Blood. 2011 Mar 10;117(10):2778–2782.
- Hervier B, Arnaud L, Charlotte F, et al. Treatment of Erdheim-Chester disease with long-term high-dose interferon-α. Semin Arthritis Rheum. 2012 Jun;41(6):907–913. Epub 2012 Jan 31.
- Diamond EL, Abdel-Wahab O, Pentosova E, et al. Detection of an NRAS mutation in Erdheim-Chester disease. Blood. 2013;122(6):1089–1091.
- Dagna L, Corti A, Langheim S, et al. Tumor necrosis factor α as a master regulator of inflammation in Erdheim-Chester disease: Rationale for treatment with infliximab. J Clin Oncol. 2012 Oct 1;30(28):e286–e290.
- Ferrero E, Belloni D, Corti A, et al. TNF-α in Erdheim-Chester disease pericardial effusion promotes endothelial leakage in vitro and is neurtralized by infliximab. Rheumatology (Oxford). 2014 Jan;53(1):198–200.
- Aouba A, Georgin-Lavialle S, Pagnoux C, et al. Rationale and efficacy of interleukin-1 targeting in Erdheim-Chester disease. Blood. 2010 Nov 18;116(20):4070–4076.
- Killu AM, Liang JJ, Jaffe AS. Erdheim-Chester disease with cardiac involvement successfully treated with anakinra. Int J Cardiol. 2013 Sep 1;167(5):e115–e117.
- Darsetin F, Kirschey S, Heckl S, et al. Successful treatment of Erdheim-Chester disease with combination of interleukin-1-targeting drugs and high-dose glucocorticoids. Intern Med J. 2014 Jan;44(1):90–92.
- Tran T, Pariente D, Guitton C, et al. Treatment of Erdheim-Chester disease with canakinumab. Rheumatology (Oxford). 2014 Dec;53(12):2312–2315.
- Haroche J, Cohen-Aubart F, Emile JF, et al. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAFV600-Mmutated Erdheim-Chester diseaese. J Clin Oncol. 2015 Feb 10;33(5):411–418.
- Houston BA, Miller PE, Rooper LM, et al. Clinical problem solving. From dancing to debilitated. N Engl J Med. 2016 Feb 4;374(5):470–477.
- Singh JA, Beg S, Angeles Lopez M. Tocilizumab for rheumatoid arthritis: A Cochrane systematic review. J Rheumatol. 2011 Jan;38(1);10–20.
- Berti A, Ferrarini M, Ferrero E, et al. Cardiovascular manifestations of Erdheim-Chester disease. Clin Exp Rheumatol. 2015 Mar–Apr;33(2 Suppl 89):S155–S163.