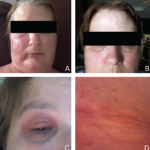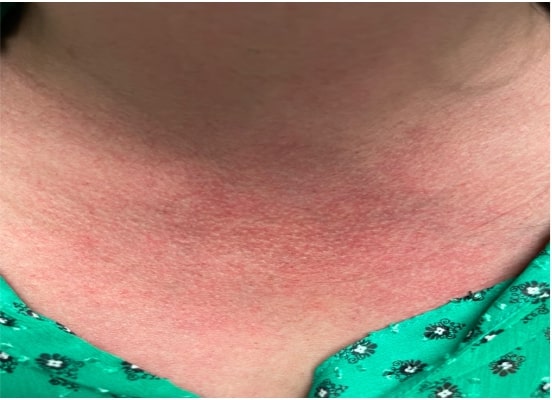
FIGURE 1: Cutaneous flushing and patchy erythematous macules on the neck and upper chest.
Systemic autoinflammatory diseases (SAIDs) are rare syndromes characterized by alterations in innate immunity that result in a variety of clinical manifestations that are usually associated with recurrent fevers.1 Thanks to advances in genetic sequencing over the past few years, monogenic causes for some of these autoinflammatory diseases, such as Yao syndrome, have been discovered.2
Previously known as nucleotide-binding oligomerization domain 2 (NOD2) associated autoinflammatory disease, Yao syndrome is characterized by periodic fever, gastrointestinal manifestations, dermatitis, polyarthritis and sicca-like symptoms. The disease occurs sporadically in most patients, and a family history is seen in only a few.3
We report the case of a patient diagnosed with Yao syndrome who was discovered to have two genetic mutations associated with autoinflammatory disease, as well as a COVID-19 infection that may have contributed to her final clinical presentation.
Case Description
A 46-year-old woman of both Asian and Latin descent presented with a chief complaint of recurrent episodic fevers associated with abdominal pain and non-bloody diarrhea for the prior 12 years.
Initially these episodes lasted for several days and occurred infrequently, months to years apart. The abdominal pain was generalized with mild nausea and occasional vomiting during these episodes. Upon further questioning, the patient reported swelling along her hands, ankles and feet, with a concomitant rash and facial flushing during these episodes. The flushing was reported to be related to heat exposure, and the rash was described as non-pruritic, extending over her face and chest, and persisting up to several weeks at times.
The patient presented to our clinic only after the episodic nature of her symptoms appeared to transition into a more persistent pattern.
Apart from fatigue and mild sicca symptoms, a review of the patient’s systems was otherwise unremarkable. Pertinent negatives included lack of weight loss, night sweats, alopecia, ocular complaints, oral ulcers, parotid gland enlargement, cough, shortness of breath, chest pain, myalgias, muscle weakness or Raynaud’s phenomenon.
She had a past medical history of hypertension, vitamin D deficiency and polycystic ovarian syndrome. Her medications included lisinopril, hydrochlorothiazide and vitamin D. The patient’s surgical history included only a remote history of tonsillectomy. She reported an allergic reaction to sulfa antibiotics resulting in a rash and laryngeal edema.
In regard to her social history, she had no history of smoking, alcohol use or illicit drug use. She worked as an elementary school teacher and was able to perform her basic activities of daily living (ADLs) without any limitations. The patient denied any notable medical diagnoses in her three children apart from a history of IgA vasculitis in one of her sons. Her father was born in China, and her maternal family originated from Cuba. She denied any history of travel and did not own any pets. There was no known family history of periodic fever syndromes.
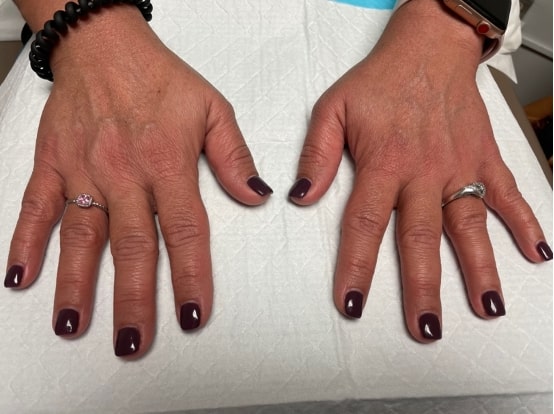
FIGURE 2: Synovitis of the second and fourth proximal interphalangeal joints.
On physical exam, the patient’s vital signs were within normal limits, with a temperature of 98.9ºF. The only notable findings were an erythematous rash along her upper chest (see Figure 1) and synovitis of both the second and fourth proximal interphalangeal joints (see Figure 2). She had no palpable lymphadenopathy, parotid enlargement, conjunctivitis or oral ulcers. The cardiopulmonary examination was unremarkable. An abdominal examination revealed a non-tender abdomen without distention, rebound or hepatosplenomegaly. Muscle strength testing was normal, and she had no neurological deficits.
Testing in 2009 ruled out an infectious etiology; the investigation was largely unremarkable apart from a positive Epstein-Barr virus (EBV) IgG titer. Further evaluation was halted until the patient had a recurrence of her episodes four years later, when she again underwent extensive testing for an infectious cause of her symptoms, this time at an academic medical center. Stool cultures, blood cultures, viral hepatitis, HIV, QuantiFERON-TB, parvovirus, testing for Lyme disease, rickettsia, C. difficile, blastomycosis and histoplasmosis all came back negative. The patient’s complete blood count, comprehensive metabolic panel, erythrocyte sedimentation rate, C-reactive protein and urinalysis were all within normal limits on multiple occasions. Other tests, such as creatinine kinase, aldolase, thyroid-stimulating hormone (TSH), serum protein electrophoresis and serum immunoglobulins, came back normal, as well. Testing for celiac disease was negative. A rheumatologic evaluation was negative for anti-nuclear antibodies, including any extractable nuclear antigens, anti-neutrophil cytoplasmic antibodies (ANCA), rheumatoid factor, anti-cyclic citrullinated peptide (CCP) antibody, HLA-B27 and angiotensin-converting enzyme levels.
Given that multiple differential diagnoses had been ruled out at this point, consideration was given to autoinflammatory disease as a possible diagnosis. The patient was subsequently prescribed 0.6 mg of colchicine twice a day without any improvement.
During this time, the patient developed COVID-19 and started to notice that her periodic episodes were becoming more persistent. Concern for an underlying malignancy prompted evaluation with colonoscopy and computerized tomography (CT) scan of the chest, abdomen and pelvis, which failed to reveal any significant findings apart from a small incidental lung nodule. Further investigation for underlying autoinflammatory disease was eventually performed with Invitae genetic testing.
The genetic panel for autoinflammatory syndromes was positive and heterozygous for a mutation in the NOD2 gene, IVS8+158 and R702W variant. Interestingly, the patient was also found to be positive and heterozygous for a mutation in TNFRSF1A R92Q, the gene associated with TNF receptor-associated periodic syndrome (TRAPS).
Despite having mutations associated with two autoinflammatory disease, the patient was diagnosed with only one autoinflammatory disease, Yao syndrome, based on her clinical presentation. It was decided to start the patient on 150 mg of canakinumab every two months to treat her autoinflammatory disease, to which she had a good response.
Discussion
Diagnosing SAIDs can be an arduous process because the conditions often share identical clinical phenotypes and their presentations often mimic infectious, malignant, allergic and autoimmune disorders, particularly early in the course of disease. A broad differential diagnosis is invariably required and should also include the newly discovered group of autoinflammatory disorders.5,6
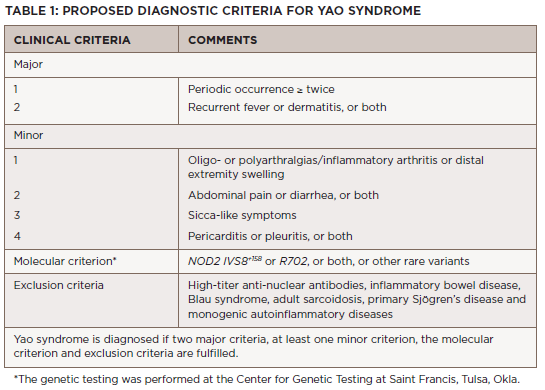
The diagnosis of Yao syndrome requires the presence of clinical criteria, consisting of typical symptoms; molecular criteria, consisting of NOD2 variants; and the proper exclusion of similar diseases, such as inflammatory bowel syndrome, sarcoidosis, Sjögren’s disease, Blau syndrome and other monogenic autoinflammatory syndromes.3,7 The NOD2 variant IVS8+158 is detected in almost all patients with Yao syndrome, with concurrent R702W in up to 30%.3,7
Click to enlarge.
Recently, our growing understanding of the disease has identified even newer genetic variants and other unique manifestations, such as eyelid swelling.8 Table 1 shows the proposed diagnostic criteria for Yao syndrome.9
Differentiating Yao syndrome from other hereditary periodic fever syndromes is an essential part of the diagnostic evaluation.4 Of the autoinflammatory disorders, familial Mediterranean fever (FMF) appears to have the closest phenotypic overlap with Yao syndrome despite having its own distinct genetic mutation in the MEFV gene.
Notable distinguishing features of FMF include a shorter duration of febrile episodes, usually lasting less than 72 hours, and lower extremity predilection for the rash. In contrast, Yao syndrome is characterized by predominantly facial and upper extremity rash, and the development of constipation, with some patients presenting with acute abdomen. Attempts to treat with colchicine can be revealing because patients with Yao syndrome don’t typically respond to, whereas it is the drug of choice for the treatment of patients with FMF.
The diagnosis of FMF was ruled out in our patient due to negative genetic testing for a mutation in the MEFV gene and lack of improvement with a prior trial with colchicine. Our choice of treatment with canakinumab was based on prior reports showing effective treatment with interleukin 1β neutralization.10 Other options include glucocorticoids, which can be given in pulses, and sulfasalazine, to which our patient was allergic.
Interestingly, there have been case reports of patients with concurrent mutations in both genes presenting as either an isolated Yao syndrome or an amalgamation between FMF and Yao syndrome.4 Although our patient had symptoms of only Yao syndrome, genetic testing in our patient revealed a mutation in both NOD2 and in the TNFRSF1A gene, associated with TRAPS.
Apart from the fever and abdominal pain present in both syndromes, TRAPS is associated with the development of localized myalgias, with overlying skin eruptions, pleurisy and periorbital edema, all of which were absent in our patient. Further, patients with TRAPS usually report a family history of the disease.5,11
The finding of more than one mutation associated with autoinflammatory disease in our patient raises questions about how these mutations interact with one another. Although clinically our patient fits most closely with the NOD2 mutation, there is uncertainty regarding the prognostic and management implications of the TNFRSF1A gene mutation in a patient with phenotypic Yao syndrome.
The pathogenesis of Yao syndrome remains elusive. NOD2 is a cytosolic innate immune sensor, working to maintain immune homeostasis as well as to monitor the environment for pathogenic material such as the peptidoglycan contained within the cell wall of bacteria. These features of NOD2 make it critical in the regulation of the immune mediated inflammatory response. Mutations in NOD2 have been implicated in Crohn’s disease, Blau syndrome, and most recently in Yao syndrome. The pathogenesis of Yao syndrome is considered to be multifactorial, dependent on the interplay between NOD2 function, other innate immune sensor mutations and the environment.5,12
As an example of the implications of this intricate network, a recent case report revealed the development of VEXAS syndrome, one of the newly discovered autoinflammatory conditions, after SARS-CoV-2 infection in a patient with both NOD2 and UBA1 gene mutations.13 The mechanisms responsible for this presentation are not well defined but appear to involve loss of the normal ubiquitination of the NOD2 sensor by genes such as UBA1, further enhanced by SARS-CoV-2’s innate deubiquitinating enzymes. An additional component to the pathogenesis lies in the fact that ssRNA of SARS-CoV-2 is itself a target for activation of the NOD2 sensor.
Given that our patient presented with symptoms of her disease before her SARSCoV-2 infection, it is possible the timing of her COVID-19 illness and subsequent increased frequency of her symptoms are merely related through synchronicity. We wonder, however, if COVID-19 may have played a more direct rather than contributory role as described in other case reports because it appears to be involved in a key mechanistic pathway between cytosolic immune sensing and a dysfunctional inflammatory response.
As our understanding of autoinflammatory disease grows, we need to be aware of the possibility that a patient may harbor more than one genetic mutation, with possible phenotypic and therapeutic implications on their disease. Lastly, it is possible that infections may augment the course of these diseases and help unravel the mechanisms associated with pathogenesis.
Saud Abaalkhail, MD, is a rheumatology fellow at Larkin Community Hospital, Miami. He completed his internal medicine residency training at Saint Vincent Hospital in Massachusetts.
Muhammed Umair Javaid, DO, is a recent graduate of the rheumatology fellowship program at Larkin Community Hospital, Miami, and is now an assistant professor of medicine in the rheumatology department at Augusta University, Georgia.
Amarie Negron Rodriguez, MD, is a private practice rheumatologist in Fort Lauderdale, Fla., and is actively involved in the education and training of future rheumatologists.
Acknowledgment
The authors thank Qingping Yao, MD, PhD, both for his contributions to this article, and for expanding our knowledge and understanding of this rare condition.
References
- Yao Q, Furst DE. Autoinflammatory diseases: An update of clinical and genetic aspects. Rheumatology (Oxford). 2008 Jul;47(7):946–951.
- Yao Q, Su LC, Tomecki KJ, et al. Dermatitis as a characteristic phenotype of a new autoinflammatory disease associated with NOD2 mutations. J Am Acad Dermatol. 2013 Apr;68(4):624–631.
- Yao Q, Shen B. A systematic analysis of treatment and outcomes of NOD2-associated autoinflammatory disease. Am J Med. 2017 Mar;130(3):365.e13–365.e18.
- Yao Q, Shen M, Gorevic P. NOD2 versus MEFV: Differential diagnosis of Yao syndrome and familial Mediterranean fever. Rheumatol Immunol Res. 2021 Dec 31;2(4):233–239.
- Yao Q, Gorevic P. (November, 2020). Yao syndrome: A case report & clinical review. The Rheumatologist. 2020 Nov 12.
- Gattorno M, Hofer M, Federici S, et al. Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis. 2019 Aug;78(8):1025–1032.
- Yao Q, Shen M, McDonald C, et al. NOD2-associated autoinflammatory disease: A large cohort study. Rheumatology (Oxford). 2015 Oct;54(10):1904–1912.
- Yao Q, Kontzias A. Expansion of phenotypic and genotypic spectrum in Yao syndrome: A case series. J Clin Rheumatol. 2022 Jan 1;28(1):e156–e160.
- Yao Q, Shen B. A systematic analysis of treatment and outcomes of NOD2-associated autoinflammatory disease. Am J Med. 2017 Mar;130(3):365.e13–365.e18.
- Yao Q. Research letter: Effectiveness of canakinumab for the treatment of Yao syndrome patients. J Am Acad Dermatol. 2019 Sep 18:S0190–9622(19)32757–4.
- Yao Q, Englund KA, Hayden SP, et al. Tumor necrosis factor receptor associated periodic fever syndrome with photographic evidence of various skin disease and unusual phenotypes: Case report and literature review. Semin Arthritis Rheum. 2012 Feb;41(4):611–617.
- Yao Q. Nucleotide-binding oligomerization domain containing 2: Structure, function, and diseases. Semin Arthritis Rheum. 2013 Aug;43(1):125–130.
- Rivera EG, Patnaik A, Salvemini J, et al. SARS-CoV-2/COVID-19 and its relationship with NOD2 and ubiquitination. Clin Immunol. 2022 May;238:109027.