We present the case of a patient with a seven-year history of paroxysmal bouts of a large-joint asymmetric arthritis who, after an extensive workup, was found to be heterozygous for the K695R mutation in pyrin.
The Case
A 33-year-old Caucasian man with a past medical history of hiatal hernia and peptic ulcer disease was referred to rheumatology for recurrent bouts of acute onset knee pain and swelling. The patient reported he was in his usual state of health until six to seven years earlier when he noticed the insidious onset of worsening arthralgias. Arthralgias began in his knuckles bilaterally and, over the course of several years, affected all the large and small joints of his upper and lower extremities. The pain was described as non-radiating, moderate to severe and aching in nature, with an occasional stabbing sensation. His symptoms were partially controlled with acetaminophen. The patient noted that activity worsened his pain, but alleviated his stiffness. Morning stiffness was reported as lasting two to three hours daily.
His family history was significant for his father dying at age 57 from hepatitis C-related cirrhosis and his mother dying of an unknown primary cancer at age 42. He previously smoked two packs of cigarettes a day but had quit 14 years earlier. Surgical history was significant for cholecystectomy, which was performed for recurrent bouts of abdominal pain. His initial physical exam demonstrated no evidence of synovitis in his large or small joints beyond tenderness involving the right fourth proximal interphalangeal joint. He had several serologic studies that started the workup noted in Table 1, all of which were negative. X-rays taken of his chest and hands at his initial evaluation proved unremarkable.
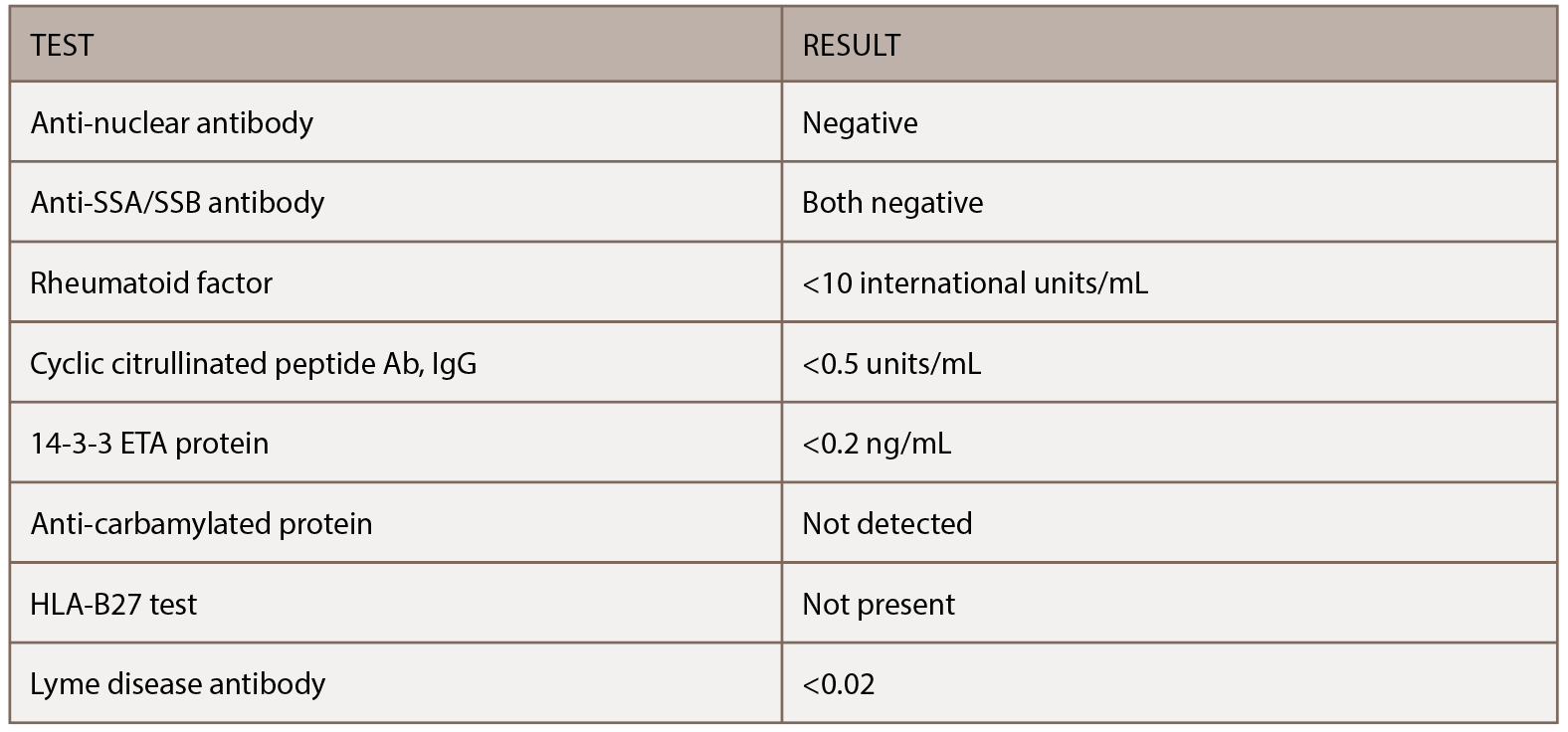
(click for larger image) Table 1: Serologic Study Results
One month later, the patient presented for follow-up, two weeks after rapid onset of swelling in both knees. Symptoms started rapidly and ended abruptly two to three days later. The swelling and pain affected his left knee more than the right. Examination of the left knee at this appointment demonstrated some minor swelling and suprapatellar fullness, so arthrocentesis was performed, which yielded no synovial fluid. Given the patient’s report of recurrent bouts of swelling and pain in the left knee, magnetic resonance imaging (MRI) of the left knee was ordered. This report demonstrated mild chondromalacia and a small effusion, but no other significant abnormalities. Additional serologic studies were ordered and were unrevealing.
His symptoms seemed to abate, and at follow-up four months later, he reported having low-grade intermittent flares of arthritis, but the severity had started to improve.
Nine months after his initial evaluation, the patient requested urgent evaluation. He reported the rapid onset of left knee pain and swelling, which was making weight-bearing difficult. He noted that his pain developed in the absence of an antecedent traumatic event or recent infection. He was evaluated within 48 hours and had an almost normal exam, except for slight loss of the peripatellar fossa of the left knee. He showed pictures of his knees on his smartphone taken when the left knee first became acutely inflamed (see Figure 1, left). Based on the photos, his recurrent knee symptoms and the unremarkable rheumatologic workup to date, genetic testing for a periodic fever syndrome was performed. Testing demonstrated heterozygosity for the K695R mutation in pyrin (marenostrin) protein. He was started on colchicine, and although it initially seemed beneficial, his symptoms gradually recurred.
Discussion
FMF is a rare, inherited disorder characterized by recurrent bouts of inflammation affecting the abdomen, chest or joints. Episodes last 12–72 hours, can vary in severity and are often accompanied by fever, erysipelas-like rash or headache. Although genetic testing for MEFV mutations is preferred when making an FMF diagnosis, the diagnosis remains clinical, because mutations have varying penetrance and homozygosity cannot always be demonstrated.3 FMF diagnosis can be made using the Tel-Hashomer clinical criteria, in which two or more major symptoms or one major plus two minor symptoms are present.4 (Major and minor Tel-Hashomer clinical criteria appear in Table 2) Livneh et al. suggested a version of FMF diagnostic criteria that included descriptions of typical and incomplete symptoms, as noted in Table 3 (opposite).5 As access to genetic testing for FMF expands, identification of biallelic MEFV pathogenic variants can confirm the diagnosis.
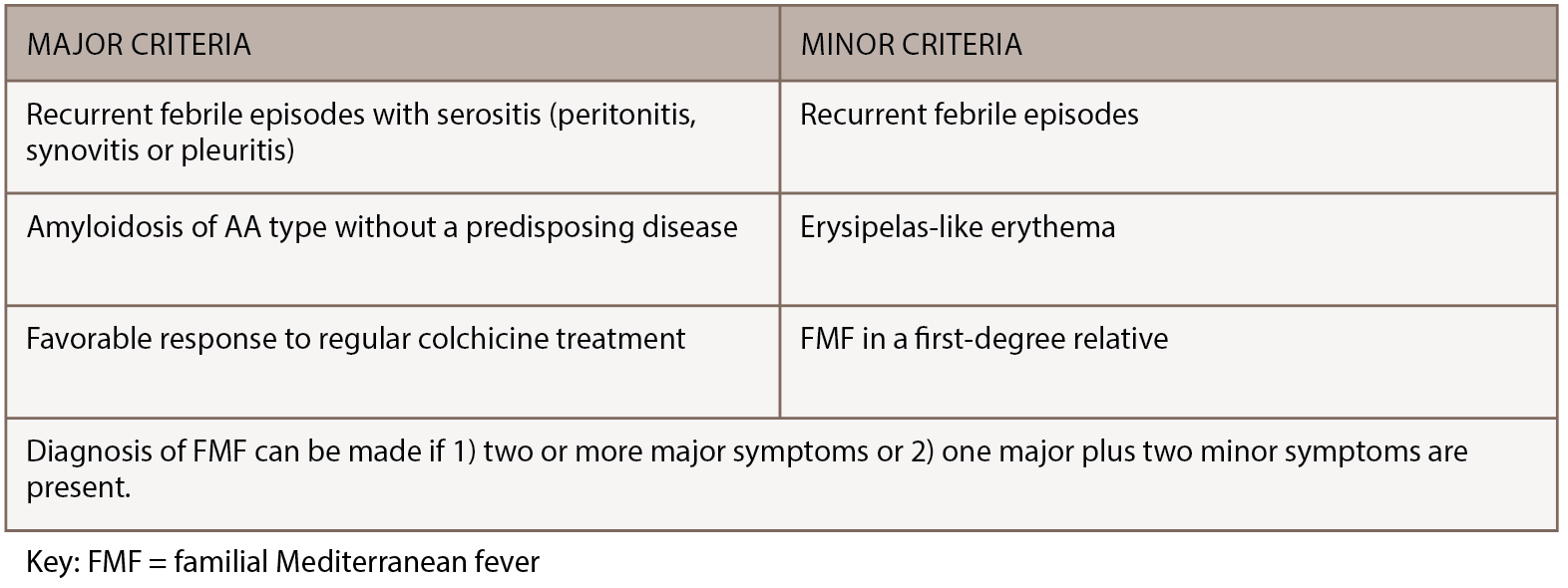
(click for larger image) Table 2: Tel-Hashomer Diagnosis Criteria
But how does the clinician make a diagnosis of FMF when atypical clinical manifestations or, as in this case, musculoskeletal predominant symptoms are the main features? For this patient, after an extensive serologic and radiographic workup for the presence of other systemic autoimmune arthritides was unrevealing, arthrocentesis and, ultimately, MRI of the knee were pursued to determine if an intra-articular etiology, such as pigmented villonodular synovitis, for his recent swelling was present. These studies were also unrevealing. It wasn’t until the patient showed photos of his knees on his smartphone to the rheumatologist that a break in the case occurred. Ultimately, it was a combination of persistence by both the patient and the physician, as well as the ability to evaluate the patient expeditiously, that suggested he might have an autoinflammatory disorder.
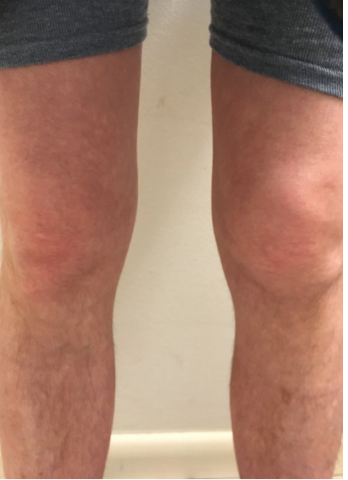
Figure 1: The patient provided photos of both knees. Note the swelling in suprapatellar bursa and loss of medial/lateral peripatellar fossa.
How does the clinician proceed with establishing a diagnosis of FMF when only one allelic variant is present? Although FMF is typically described as an autosomal recessive disorder, it is becoming increasingly well known that up to 30% of patients with FMF have only one detected pathogenic MEFV variant despite sequencing of the entire coding region. These patients frequently have a typical clinical history and respond well to colchicine.2
Multiple reasons for this phenomenon exist. First, some routine genetic screening tests might target only the most prevalent mutations, so novel or rare mutations might be missed.2 Indeed, several mutations (i.e., E148Q on exon 2 and M680I, M694I, M694V and V726A on exon 10) may be responsible for up to 80% of FMF cases in classically affected populations.2
Additionally, the second mutation lies in intronic or regulatory regions (perhaps affecting messenger RNA) and thus isn’t screened for unless the entire gene was sequenced. Third, a few disease-associated mutations are gene rearrangements, such as a deletion, and not the typical missense nucleotide changes routinely screened for.2 Fourth, several pedigrees have been described with family members affected in an autosomal dominant pattern.6,7
Finally, the presence of one or several modifying genes may produce a clinical picture similar to FMF through their modulation of the innate immune response.3 Evidence exists suggesting that major histocompatibility complex class I, such as human leukocyte antigen (HLA‑A) and class II alleles, exert a differential effect on the clinical picture and response to colchicine in a Japanese population.3 The influence of these alleles may lead to the achievement of an inflammatory dosage threshold necessary for the systemic inflammation and ultimate presentation of FMF.2
In this patient, not all of the typical symptoms of FMF were present, but after several visits and a frustrating, unrevealing workup for a systemic autoimmune or inflammatory arthritis, the patient presented with a picture that clearly documented swelling in his knee. He was evaluated expeditiously and found 48–72 hours later to have had a substantial amount of improvement in his swelling. At this point, an autoinflammatory etiology was entertained.
Testing for FMF yielded heterozygosity for the K695R mutation in pyrin. Although this is a fairly obscure mutation, it is becoming increasingly recognized as associated with a severe clinical phenotype and associated with frequent bouts of arthritis, even when heterozygous.8,9 At a molecular level, although both lysine (K) and arginine (R) are basic amino acids with positively charged side chains, the change would be expected to alter protein function.10
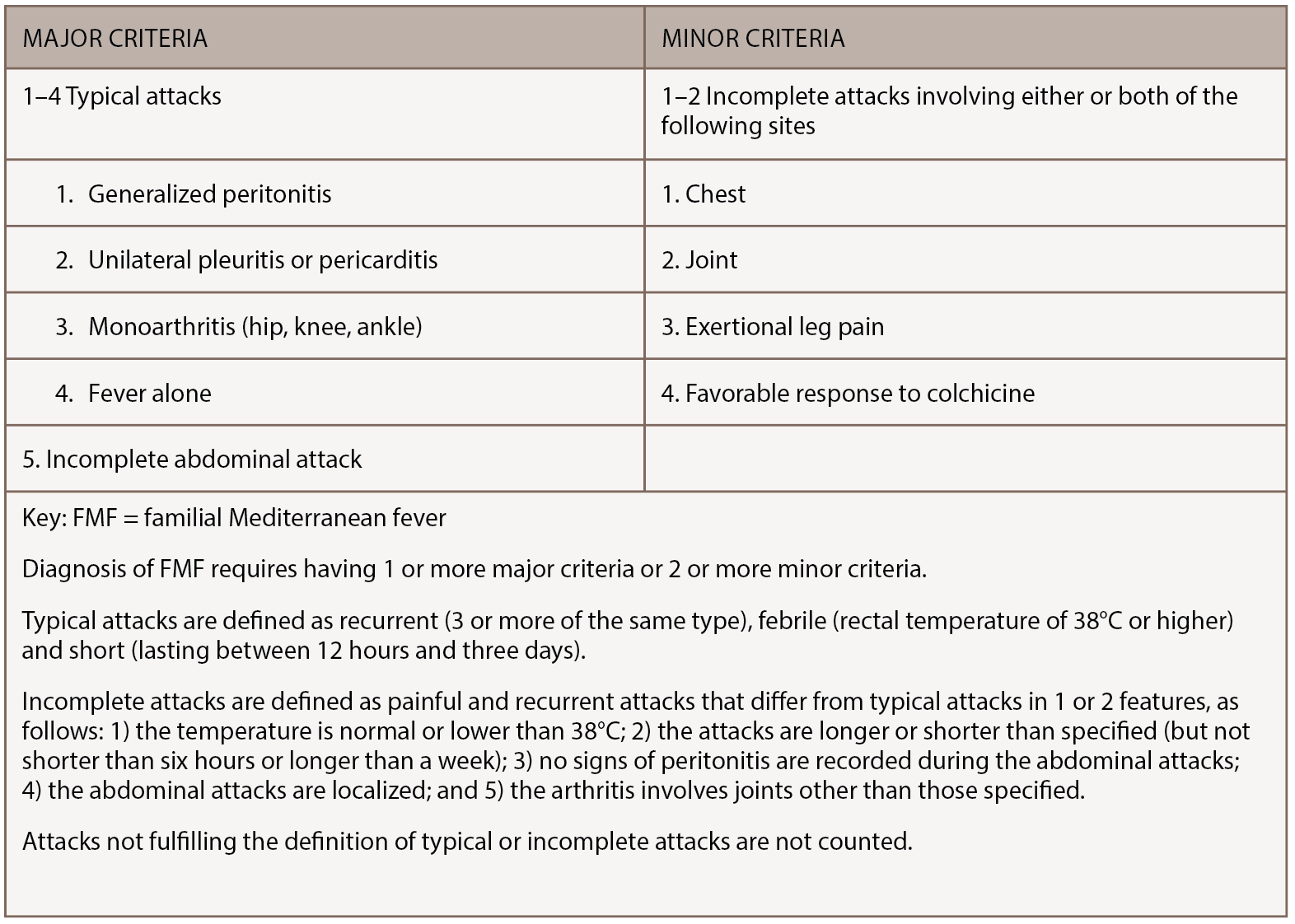
(click for larger image) Table 3: Simplified FMF Diagnosis Criteria5
Additionally, the K695R mutation, as well as multiple other clinically significant mutations in exon 10 (coding amino acids 577–781), alters the B30.2 domain, which interacts with caspase-1.11 This region is an important functional site for pyrin because it regulates the interaction with caspase-1. FMF mutations involving exon 10 represent a gain of function and, in a pro-inflammatory manner, enhance the interaction with caspase-1 to cleave precursor forms of IL-1β and, thus, unleash the inflammatory response and regulation of apoptosis.3 Pyrin also has an anti-inflammatory role, promoting autophagy of cytoplasmic regulators of innate immunity, eventually promoting the regulation of the inflammatory response.3
As noted, we started the patient on colchicine, and after several weeks he had a transient, partial response.
At his follow-up appointment after his diagnosis, we discussed alternative therapies should colchicine not control his symptoms, although the patient was encouraged to continue his colchicine for at least six months before considering it a therapeutic failure. If symptoms don’t sufficiently respond to colchicine, we may implement a trial of anakinra or a different IL-1 inhibitor. It has been observed that 5–10% of patients may not respond (or are intolerant) to colchicine, so close clinical monitoring for a durable response is important.3
Although this case highlighted many different teaching points to the rheumatologist providing his care, it was ultimately patience—on the part of both the patient and his doctor—along with telltale photos that led to his diagnosis.
 Taylor T. Faulk, MD, is an internal medicine resident at Keesler Medical Center at Keesler Air Force Base in Mississippi.
Taylor T. Faulk, MD, is an internal medicine resident at Keesler Medical Center at Keesler Air Force Base in Mississippi.
 Matthew B. Carroll, MD, FACP, FACR, is a practicing rheumatologist at the Singing River Health System in Pascagoula, Miss.
Matthew B. Carroll, MD, FACP, FACR, is a practicing rheumatologist at the Singing River Health System in Pascagoula, Miss.
References
- Ciccarelli F, De Martinis M, Ginaldi L. An update on autoinflammatory diseases. Curr Med Chem. 2014;21(3):261–269.
- Booty MG, Chae JJ, Masters SL, et al. Familial Mediterranean fever with a single MEFV mutation: Where is the second hit? Arthritis Rheum. 2009 Jun;60(6):1851–1861.
- Manukyan G, Aminov R. Update on pyrin functions and mechanisms of familial Mediterranean fever. Front Microbiol. 2016 Mar 31;7:456.
- Pras M. Familial Mediterranean fever: From the clinical syndrome to the cloning of the pyrin gene. Scand J Rheumatol. 1998;27(2):92–97.
- Livneh A, Langevitz P, Zemer D, et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 1997 Oct;40(10):1879–1885.
- Booth DR, Gillmore JD, Lachmann HJ, et al. The genetic basis of autosomal dominant familial Mediterranean fever. QJM. 2000 Apr;93(4):217–221.
- Aldea A, Campistol JM, Arostegui JI, et al. A severe autosomal-dominant periodic inflammatory disorder with renal AA amyloidosis and colchicine resistance associated to the MEFV H478Y variant in a Spanish kindred: An unusual familial Mediterranean fever phenotype or another MEFV-associated periodic inflammatory disorder? Am J Med Genet A. 2004 Jan 1;124A(1):67–73.
- Cekin N, Akyurek ME, Pinarbasi E, et al. MEFV mutations and their relation to major clinical symptoms of familial Mediterranean fever. Gene. 2017 Aug 30;626:9–13.
- Battal F, Silan F, Topaloğlu N, et al. The MEFV gene pathogenic variants and phenotype-genotype correlation in children with familial Mediterranean fever in the Çanakkale population. Balkan J Med Genet. 2017 Mar 5;19(2):23–28.
- Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010 Apr;7(4):248–249.
- Moradian MM, Babikyan D, Banoian D, et al. Comprehensive analysis of mutations in the MEFV gene reveal that the location and not the substitution type determines symptom severity in FMF. Mol Genet Genomic Med. 2017 Nov;5(6):742–750.


