His symptoms, swelling and pain in his hands, wrists, ankles and toes, began nine days after he had a root canal and crown preparation. He went to an emergency department, where it was noted he had had a tick exposure. He was prescribed doxycycline and discharged.
Click to enlarge.
Two weeks later, he presented to the same emergency department with a fever of 103.4º and was admitted for suspected sepsis. He had an extensive evaluation, including blood cultures, a tickborne illness panel, computed tomography (CT) scans of his chest, neck, abdomen and pelvis, and transthoracic cardiac echocardiogram, which showed no acute abnormalities. Laboratory tests and results from that presentation are listed in Table 1.
Additional testing included:
- Serum protein electrophoresis (SPEP): consistent with acute phasereactant, faint restricted-band M spike migrating in the gamma-globulin region;
- Immunofixation: A faint band in IgG kappa could not be ruled out from the serum immunofixation pattern;
- Parvovirus B-19 IgM: negative at 0.1 (cutoff <0.9);
- Parvovirus B-19 IgG: elevated, positive at 4.3; (cutoff <0.9);
- Anti-neutrophilic cytoplasmic antibody (ANCA): negative;
- Hepatitis B and C naive by antibody testing;
- HIV: antibody negative;
- Parvovirus B19 IgM: negative, protein C: normal, protein S: normal;
- Factor 5 Leiden: normal;
- Lupus anticoagulant (dilute Russell viper venom time): 1.19 (cutoff <1.14);
- Anti-centromere antibody: negative;
- Anti-nuclear antibody (ANA): 1:80;
- Double-stranded DNA: negative;
- Rheumatoid factor: negative;
- anti-RNP and anti-Smith antibodies: negative;
- anti-Sjögren’s antibodies: negative;
- Cold agglutinins: negative;
- Urinalysis: normal;
- Legionella antigen: negative;
- Streptococcus pneumonia antigen: negative;
- Epstein Barr virus (EBV) nuclear antibody IgG: positive (cutoff >21.99);
- EBV capsid antibody IgM: negative;
- Cytomegalovirus (CMV) antibody IgM: negative;
- Anaplasma phagocytophilum DNA: not detected;
- Babesi microti DNA: not detected;
- Borrelia miyamotoi DNA: not detected;
- Ehrlichia chaffeensis DNA: negative;
- Lyme disease DNA: not detected.
He was seen by an infectious disease specialist who believed the patient had a viral illness. The patient was discharged.
The patient’s fevers resolved; however, his arthralgias persisted. In addition, he developed cyanotic changes in his fingertips and toes. In follow-up with his primary care physician a few days later, he was started on 40 mg of prednisone daily to treat his elevated ANA.
He returned to his dentist, who treated him with fluconazole for oral thrush, and his arthralgias improved after a few days.
He returned to the hospital one week later for acrocyanosis. He underwent vascular studies, including venous Doppler ultrasounds, which were negative for deep vein thrombosis (DVT), and spiral CT to rule out pulmonary embolism. Pulse volume recording (PVR) and anklebrachial index (ABI) tests showed normal triphasic Dopplers and wave forms, except in his toes. CT angiography with runoff showed patent major arteries from the aorta to the ankles, without stenosis.
He was evaluated by a vascular surgeon, who ruled out macrovascular disease. He was started on procardia and nitropaste for suspected thromboangiitis obliterans (TAO) and was advised to stop smoking.
Two weeks later, he was seen again by his primary care physician, who increased the prednisone to 60 mg daily, which gave him partial relief from the squeezing pains in his feet and hands. The patient was referred to a rheumatologist.
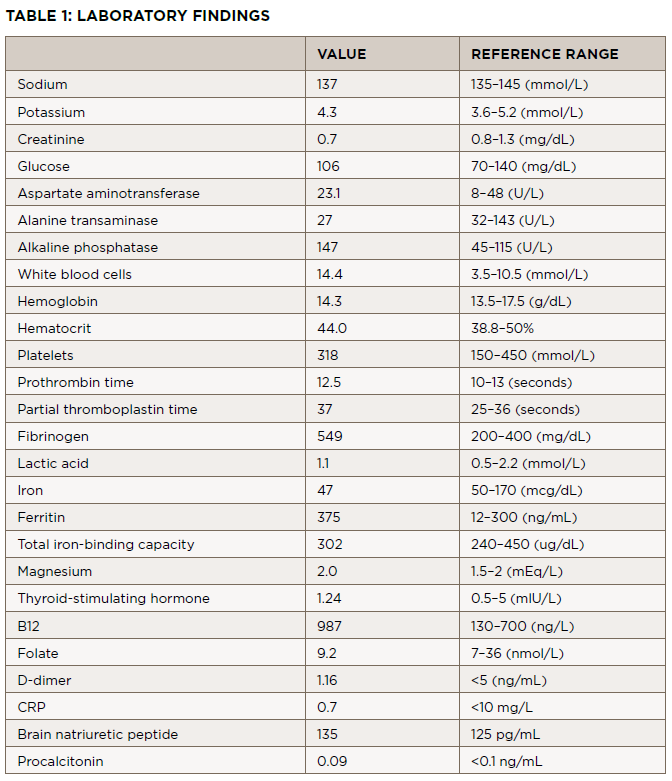
FIGURE A: Acrocyanosis, gangrene, and subungual splinter hemorrhage of the fingers before admission. FIGURE B: Acrocyanosis and gangrene of the toes before admission and prior to amputation. (Click to enlarge.)
He was seen by the rheumatologist two weeks later, who noted the patient had cyanosis with mild gangrene involving both index fingers (see Figure 1A), big toes, and his left second and third toes (see Figure 1B). Splinter hemorrhages were noted in his index fingers as well (see Figure 1A).
Despite the positive ANA, other symptoms and signs of connective tissue disease were absent, including rash, sun sensitivity, alopecia, mouth sores, pleurisy, Raynaud’s phenomenon, hemocytopenia and renal or neurologic disease. Ear, nose, throat, respiratory and renal abnormalities to suggest ANCA vasculitis were also absent. Aspirin was added to the patient’s medications, and he was continued on prednisone and nifedipine.
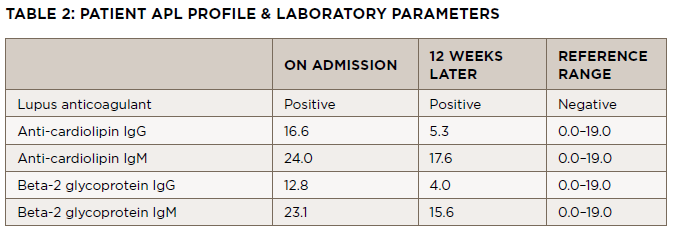
On follow-up with the rheumatologist a few days later, the ischemia in his toes had worsened, and he had developed dusky, purple patches on the soles of both feet and tissue breakdown in both index fingertips. He was in a significant amount of pain. Laboratory test results from his initial rheumatology visit included a positive aPL profile, noted in Table 2.
Click to enlarge.
Cryoglobulins, ANA, Scl-70, blood and fungal cultures were negative; complement levels were normal. His C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) had normalized.
He was admitted to our facility for possible APS and to rule out infective endocarditis in the setting of his recent dental work.
He had diminished dorsalis pedis and posterior tibial pulses, cyanotic changes in the distal tips of his toes, with blistering of toes one, two and three of his left foot. Capillary refill was extremely delayed in the affected toes, and he had ischemic changes with dark necrosis on the distal right hallux. The podiatric consultant opened the blister on the third toe, revealing clear serous fluid, which did not appear purulent. Intravenous (IV) vancomycin, IV ceftriaxone, heparin infusion, hydroxychloroquine and atorvastatin were started for limb-threatening distal ischemia in the context of his abnormal aPL profile. Blood cultures were negative, and a trans-esophageal echocardiogram was normal. The patient was discharged on warfarin, prednisone, hydroxychloroquine, nifedipine and atorvastatin.
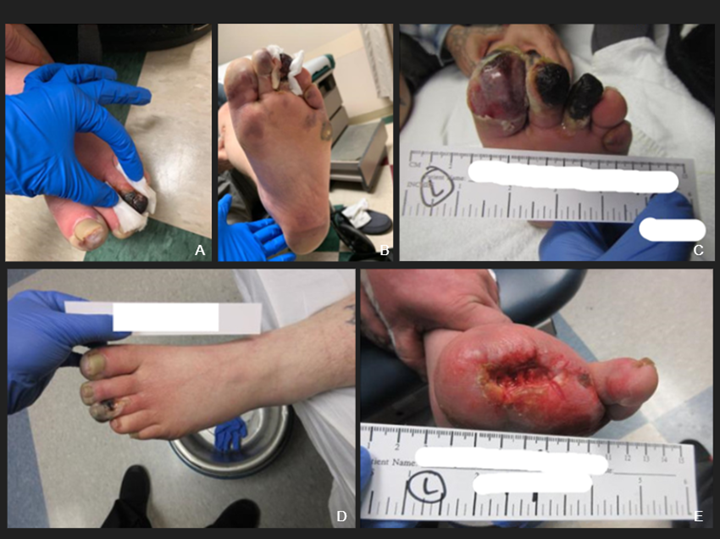
FIGURES A–C: The patient’s left foot following first hospital discharge. FIGURE D: Digital ischemia, gangrene and cellulitis of the left foot one day prior to amputation. FIGURE E: The patient’s left foot three months post-amputation. (Click to enlarge.)
The patient was seen one week after discharge. He admitted to continued smoking, despite recommendations to quit. Prednisone tapering was started in view of the normal inflammatory markers. When seen by the podiatrist a week later, he was noted to have progressive gangrene of the left first and second toes, down to bone (see Figure 2 A–D), necessitating amputation of the first, second and third toes.
Surgical pathology revealed areas of gangrenous necrosis and did not mention any signs of vasculitis. The patient was discharged on long-term antibiotics for osteomyelitis. Over the next two months, prednisone was tapered off and the other medications continued. He continued to smoke. Despite this, his digital ischemia improved and the gangrene resolved. Three months following discharge he was doing well, with no signs of recurrent disease (see Figure 2E).
An antiphospholipid panel performed exactly 12 weeks following the initial panel revealed persistent positivity of the lupus anticoagulant, with anti-cardiolipin IgM and beta-2 glycoprotein IgM levels normalizing (see Table 2).
Discussion
Digital ischemia and gangrene requiring amputation are rare and potentially lethal complications of APS. Although not included in the classification criteria for APS, dermatologic manifestations of the disorder should be recognized and appropriately evaluated. The cutaneous findings of aPL are heterogeneous, with no singular pathognomonic feature. Livedo reticularis, cutaneous ulceration and necrosis, digital gangrene and subungual splinter hemorrhages are among the dermatologic findings described in a 1,000-patient aPL cohort study by Cervera et al.3
These findings on their own are nonspecific and present a diagnostic challenge, considering the vast differential diagnosis. Evaluation is required for potential macrovascular and microvascular disease, including non-inflammatory conditions, such as thromboembolic disease, hypercoagulable states, TAO, toxin exposure and freeze injury, as well as inflammatory causes, such as infection and vasculitis.
Our patient met the classification criteria for APS as defined by Miyakis et al., including clinical episodes of thrombosis in conjunction with lupus anticoagulant on two occasions 12 weeks apart, and histopathology to exclude vasculitis.2 Imaging findings could not unequivocally confirm thrombosis because CT angiogram (CTA) with runoff did not reach beyond the patient’s ankles.
Differential Diagnosis
In our patient, who had an active smoking history and in whom macrovascular disease had been ruled out by appropriate imaging studies, TAO was a strong possibility. TAO is a diagnosis of exclusion, determined after elimination of other vascular occlusive diseases. Because cessation of smoking is the only way to prevent its progression, the improvement seen in our patient following treatment while continuing to smoke argued against this diagnosis. In addition, he had none of the classic signs of TAO in the CTA, such as a corkscrew collateral appearance of the small and medium vessels.
The hematologic evaluation includes assessment for primary (i.e., inherited) and secondary (i.e., acquired) causes of hypercoagulable states, such as Factor V Leiden and APS. In this case, the hypercoagulation panel revealed a positive lupus anticoagulant, as well as IgM cardiolipin and beta2-glycoprotein1 ABS. Twelve weeks later, the lupus anticoagulant remained positive, but the other ABS results were negative, perhaps due to treatment with glucocorticoids and hydroxychloroquine.
aPL ABS can be identified in as many as 50% of patients with systemic lupus erythematosus (SLE) and are present in 5–20% of patients with other connective tissue diseases, such as rheumatoid arthritis, dermatomyositis, Sjögren’s disease and systemic sclerosis.3 Our patient had transient joint symptoms and fever at the start of his illness, acute phase reactants were elevated, and his ANA was weakly positive. In the context of positive antiphospholipid ABS, this raised the possibility of SLE. Although SLE could not be entirely ruled out, he had no additional clinical or laboratory criteria for this condition.
Small to medium vessel vasculitis was a concern due to the prodromal fever and polyarthralgias prior to digital gangrene. However, he lacked other features of systemic vasculitis, his ANCA was negative, and vasculitis was not seen on pathology. Nevertheless, because vasculitis and lupus could not be fully ruled out, he was treated with glucocorticoids for several weeks.
Infectious etiologies of acrocyanosis and digital limb ischemia were ruled out by appropriate studies. Our patient had a history of recent dental work prior to symptom onset, which prompted evaluation for infective endocarditis with echocardiography and blood cultures. He had thrush, raising concern for candidemia, but again, blood cultures were negative. Hepatitis C-associated cryoglobulinemia was ruled out with hepatitis ABS and cryoglobulin levels.
Other less likely etiologies to be considered include chikungunya, psittacosis and mononucleosis, Coxiella burnetiid and Bartonella, but he had no travel history and did not have birds, dogs or cats at home.
Dermatologic Manifestations
Livedo reticularis, cutaneous ulceration and necrosis, digital gangrene and subungual splinter hemorrhages are among the cutaneous manifestations of APS seen by Cervaro et al. Livedo reticularis was the third most common presenting sign of APS and was prevalent at disease onset (40.4%). Only behind deep vein thrombosis (31.7%) and thrombocytopenia (21.9%), it is also present in 70% of patients with concomitant SLE and APS. The mottling of the skin seen in nonphysiologic livedo reticularis is likely due to dysfunction of blood flow resulting in the collection of deoxygenated blood between arterioles supplying adjacent (i.e.,1 to 4 cm) zones of the skin, likely from aPL autoantibodies directed at phospholipid protein complexes bound to endothelial cells within the vessels resulting in procoagulant release and thrombosis. Livedo reticularis is more commonly a physiologic response to cold exposure or hypotension; thus, on its own, it is a non-specific finding. There is no proven treatment for livedo reticularis, with low-dose aspirin often prescribed without proven efficacy.
Much less common are the manifestations of APS present in our patient, a constellation of microvascular thrombotic events that can include ulceration, necrosis and digital gangrene of the skin. Cutaneous necrosis (2.1% of patients) can be acute, painful, and widespread. These lesions may develop from peripheral erythema to necrotic bullae. Pathologic examination demonstrates non-vasculitic and non-inflammatory thrombosis of the dermal vasculature. Subungual splinter hemorrhages (0.7% of patients) are frequently associated with acute or previous thrombotic events. Digital gangrene (3.1% of patients) is a significant thrombotic event that can be caused by the blockage of large-, medium- or small-sized vessels in the limbs.
Criteria
Diagnostia criteria for APS were established by Wilson et al. in 1999.1 Since then, the body of literature regarding the disease has grown, leading to a revised consensus classification criteria for APS set forth by Miyakis et al. in 2006, with the major revisions listed in Table 3.2 The classification requires at least one clinical criterion and one laboratory criterion.
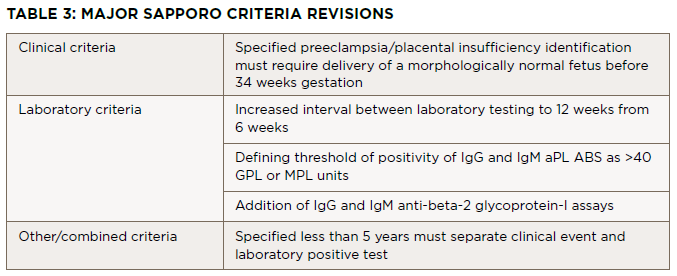
Click to enlarge.
The clinical criteria include one or more episodes of venous, arterial or small vessel thrombosis within any tissue or organ confirmed with unequivocal imaging or histopathology. The second clinical criterion is pregnancy morbidity, with three subsets of situations included. The first is the occurrence of one or more unexplained deaths of a morphologically normal fetus at or beyond the 10th week of gestation. Normality of the fetus must be confirmed by ultrasound or direct examination. The second includes one or more premature births of a normal neonate before the 34th week of gestation. This premature birth can be due to severe preeclampsia, eclampsia or placental insufficiency. The third is three or more unexplained, consecutive, spontaneous abortions before the 10th week of gestation.
Three laboratory criteria exist. First is the presence of lupus anticoagulant in plasma on two or more occasions at least 12 weeks apart. The second and third criteria are the presence of anti-cardiolipin and beta-2 glycoprotein-I antibodies respectively, (IgG or IgM isotypes) on two or more occasions at least 12 weeks apart. Miyakis et al. discussed the inclusion of skin manifestations such as livedo reticularis as independent criteria for diagnosis, but due to a lack of specificity this was rejected.2 Digital gangrene did not merit inclusion because it was considered rare.2
Treatment
The treatment of APS can be divided into primary and secondary thromboprophylaxis. Primary thromboprophylaxis is considered for aPL-positive patients who have not yet had a thrombotic event. The aim of treatment is reducing vascular risk factors, including hypercholesterolemia, obesity, smoking and use of estrogen-containing contraceptives. Although not guideline therapy, patients with high aPL titers or positivity of multiple APS-associated ABS can be treated with low-dose aspirin daily.4 Hydroxychloroquine and aspirin can be considered in patients with concomitant SLE, as well as in primary APS, because it has been demonstrated to reduce thrombosis. Statins also have proven anti-thrombotic efficacy in patients with and possible without hyperlipidemia.
Pregnant women without thrombotic events can be managed with low-dose aspirin and prophylactic-dose low molecular weight heparin (LMWH). Aspirin can also be used in the preconception period because it has been shown to aid implantation.
Secondary thromboprophylaxis is recommended for patients with proven thrombotic events. Venous thrombosis is treated in the acute setting with unfractionated heparin or LMWH, followed by chronic use of anticoagulation for a goal international normalized ratio (INR) of 2.0–3.0. Direct oral anticoagulants have not been shown to have comparable efficacy in this condition.5
Patients with arterial thrombosis or recurrent events are similarly treated with anticoagulation; however, goal INR is still uncertain because more recurrent arterial events occur with INR of 2.0–3.0, compared with venous events. Some centers recommend a goal INR of 3.0–4.0 for this group, but data are inconclusive; these patients may also be treated with combined anti-platelet and anticoagulation with warfarin with a goal INR of 2.0–3.0.6
Pregnant women with thrombotic events can be managed with therapeutic-dose LMWH, although lower doses are being used by the obstetric community with efficacy.7
As for dermatologic manifestations, low-dose aspirin and dipyridamole have been used successfully for subungual splinter hemorrhages. Digital gangrene requires full anticoagulation with heparin and/or warfarin. Iloprost and plasma exchange have been used if lesions continue to progress. Glucocorticoids and cytotoxic agents can be added in these cases for prophylaxis against aPL rebound.8
Conclusion
MORE ABOUT APS
APS should be included in the differential diagnosis when a patient presents with such dermatologic complaints as livedo reticularis. Despite not being included in the clinical criteria of APS, livedo reticularis remains the condition’s most common presenting sign. The manifestations of APS are heterogeneous, and in rare cases can include digital limb ischemia and gangrene.
Evaluation of a patient with suspected APS should include a full aPL profile, imaging and histopathology, as well as testing to exclude clinically relevant differential diagnoses. Prompt evaluation and treatment are important to prevent further thrombosis and injury.
Primary prevention of APS can include the use of aspirin, hydroxychloroquine and statins, especially in cases with concomitant SLE or hyperlipidemia, respectively. Treatment of acute thrombosis includes anticoagulation with or without antiplatelet agents, followed by long-term anticoagulation with warfarin to prevent stroke, myocardial infarction, pulmonary embolism or other complications.
 Nedal Darwish, MD, is a third-year internal medicine resident at Arnot Ogden Medical Center, Elmira, N.Y., and an incoming nephrology fellow at Baylor College of Medicine Medical Center, Houston. He has experience in clinical and academic research, having worked in microbiology and immunology, cardiology and neurology labs at NYU Langone Medical Center and SUNY Upstate Medical Center.
Nedal Darwish, MD, is a third-year internal medicine resident at Arnot Ogden Medical Center, Elmira, N.Y., and an incoming nephrology fellow at Baylor College of Medicine Medical Center, Houston. He has experience in clinical and academic research, having worked in microbiology and immunology, cardiology and neurology labs at NYU Langone Medical Center and SUNY Upstate Medical Center.
 Mohamed Manaa, MD, is a first-year internal medicine resident at Arnot Ogden Medical Center, Elmira, N.Y. He is interested in pursuing a career in rheumatology following residency.
Mohamed Manaa, MD, is a first-year internal medicine resident at Arnot Ogden Medical Center, Elmira, N.Y. He is interested in pursuing a career in rheumatology following residency.
 Griffin Reyes, MD, is an orthopedic research assistant at the University of Texas Health Science Center, Houston and an incoming preliminary surgery resident at the University of Texas Health Science Center, Houston. He is pursuing residency training in orthopedic surgery.
Griffin Reyes, MD, is an orthopedic research assistant at the University of Texas Health Science Center, Houston and an incoming preliminary surgery resident at the University of Texas Health Science Center, Houston. He is pursuing residency training in orthopedic surgery.
 James G. Freeman, MD, is a clinical assistant professor of medicine at Lake Erie College of Osteopathic Medicine and a practicing rheumatologist in upstate New York. He completed his fellowship training at the University of Pennsylvania School of Medicine, Philadelphia, and a residency in internal medicine at Boston University Medical Center.
James G. Freeman, MD, is a clinical assistant professor of medicine at Lake Erie College of Osteopathic Medicine and a practicing rheumatologist in upstate New York. He completed his fellowship training at the University of Pennsylvania School of Medicine, Philadelphia, and a residency in internal medicine at Boston University Medical Center.
References
- Wilson WA, Gharavi AE, Koike T, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: Report of an international workshop. Arthritis Rheum. 1999 Jul;42(7):1309–1311.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006 Feb;4(2):295–306.
- Cervera R, Piette JC, Font J, et al. Antiphospholipid syndrome: Clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum. 2002 Apr;46(4):1019–1027.
- Negrini S, Pappalardo F, Murdaca G, et al. The antiphospholipid syndrome: From pathophysiology to treatment. Clin Exp Med. 2017 Aug;17(3):257–267.
- Bala M, Undas A, et al. Antiplatelet and anticoagulant agents for secondary prevention of stroke and other thromboembolic events in people with antiphospholipid syndrome. Cochrane Database Syst Rev. 2020 Oct 12;10(10):CD012169.
- Rodziewicz M, D’Cruz DP. An update on the management of antiphospholipid syndrome. Ther Adv Musculoskelet Dis. 2020 Apr 27;12:1759720X20910855.
- Piette JC, Karmochkine M, Papo T, et al. Treatment of the antiphospholipid syndrome. Clin Rev Allergy Immunol. 1995 Spring;13(1):73–89.
- Lockshin MD. Anticoagulation in management of antiphospholipid antibody syndrome in pregnancy. Clin Lab Med. 2013 Jun;33(2):367–376.
