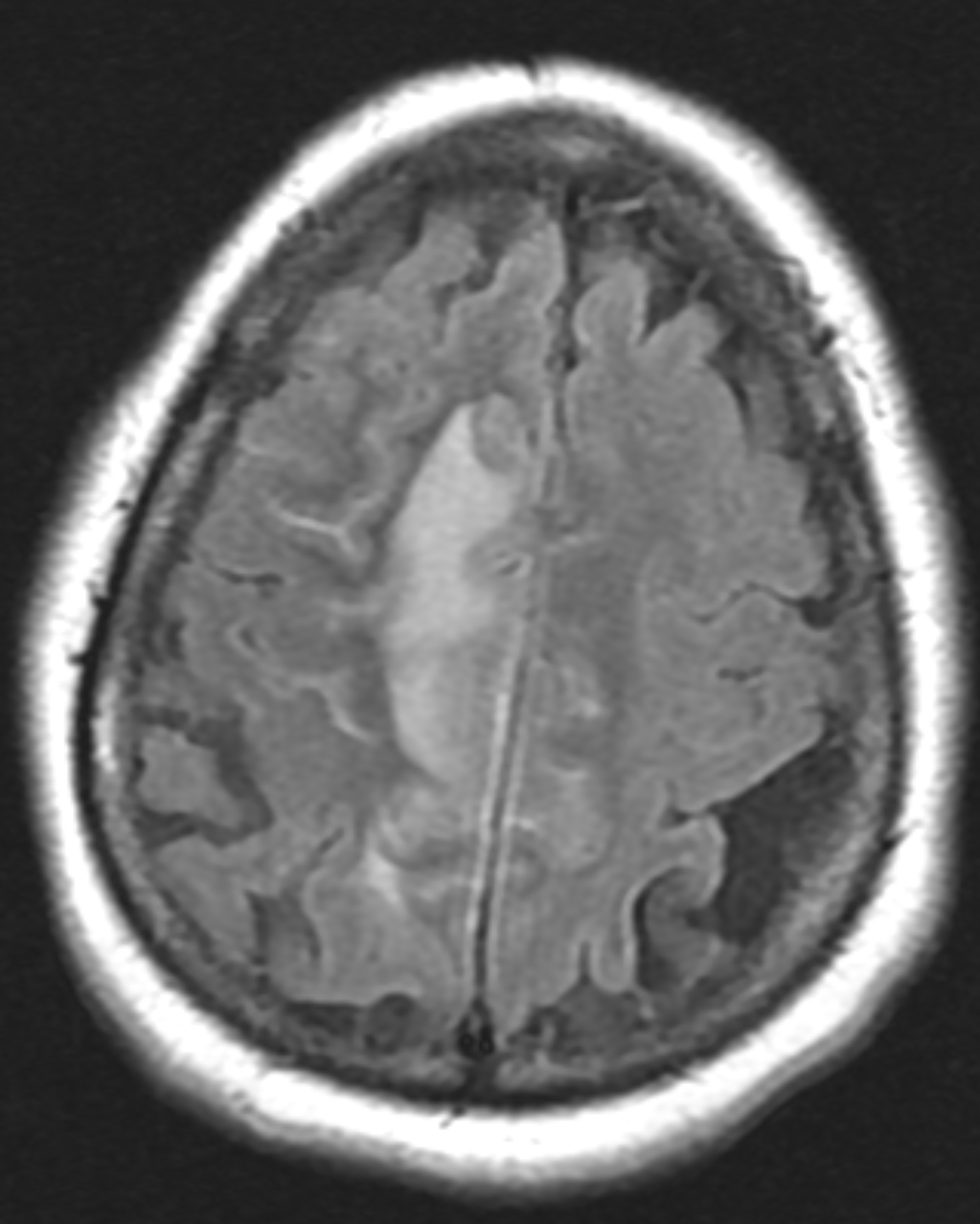
Figure 1A: T2/FLAIR sequence demonstrating right greater than left parasagittal frontal and parietal cortical T2/FLAIR hyperintensity and edema with involvement of the subcortical and deep white matter. There is also incomplete FLAIR suppression of the cerebrospinal fluid in the adjacent sulci, indicative of an inflammatory process. (Click to enlarge.)
Cerebral amyloid angiopathy (CAA) is a small-vessel disease of the central nervous system (CNS) characterized by amyloid-β deposition along the leptomeningeal and cortical vessels.1,2 Although CAA is a non-inflammatory process, an immunological response against the amyloid β peptides can develop in some patients, leading to an inflammatory, vasculitic CAA.2
Inflammatory CAA has a heterogeneous presentation sometimes mimicking CNS vasculitis, CNS infection and malignant conditions, posing a significant diagnostic challenge.3-6 The condition has two pathologically recognized subtypes: inflammatory-related CAA (CAAri), characterized by perivascular inflammatory infiltrates; and amyloid β-related angiitis (ABRA), characterized by more widespread vessel involvement with transmural, granulomatous infiltrates.7
Case Description
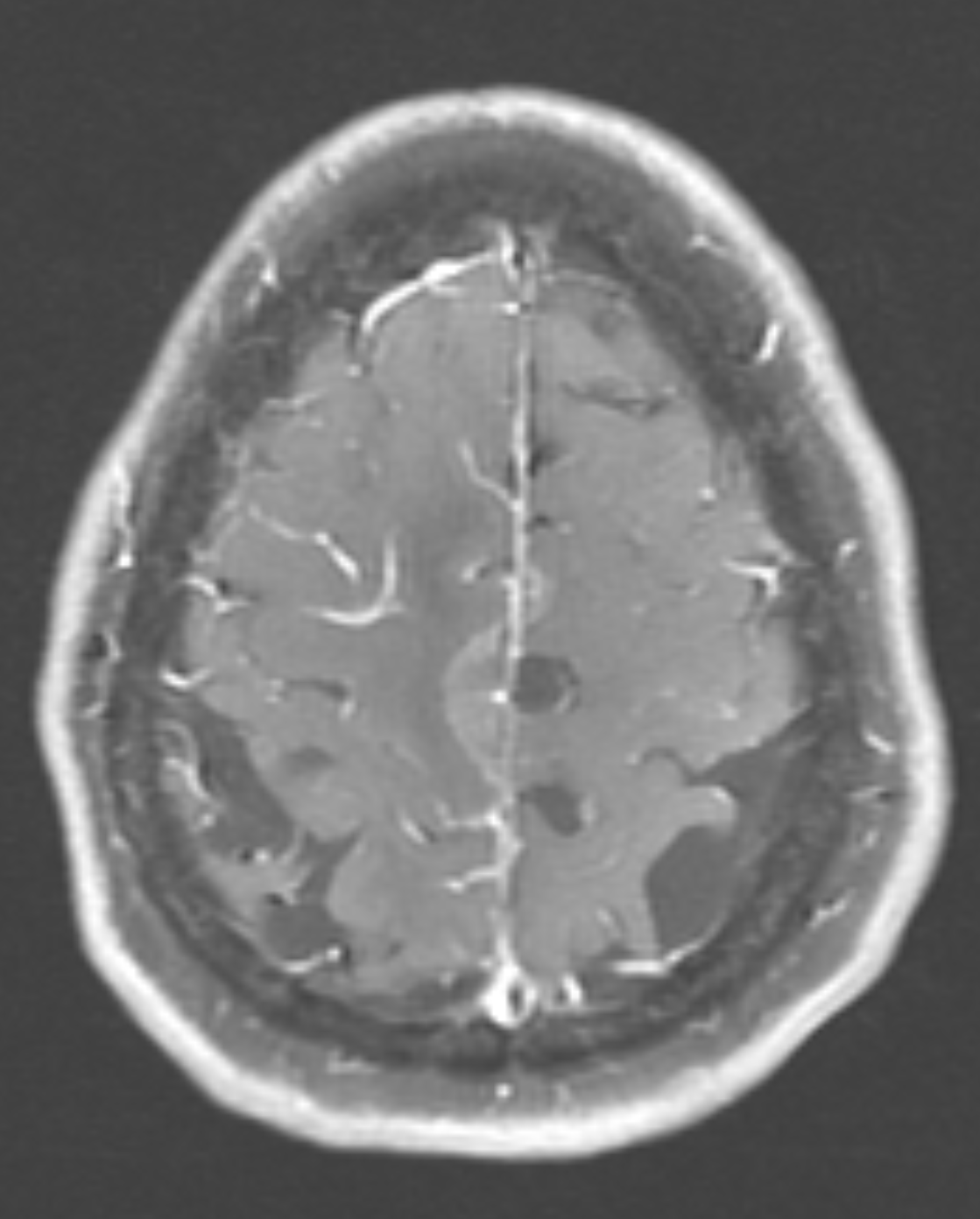
Figure 1B: T1, post contrast, showing cortical and leptomeningeal
enhancement. (Click to enlarge.)
A 72-year-old woman, with a history of hypertension, hyperlipidemia and cutaneous porphyria, presented to a local hospital with a 10-day history of headache and recurrent falls. Upon initial encounter, the physical examination was remarkable for left-sided weakness in the arm (4/5) and leg (1/5), and gait instability.
Magnetic resonance imaging (MRI) of her brain revealed a right parasagittal T2/FLAIR, and cortical and subcortical white matter hyperintensity with gyral enhancement concerning for primary or metastatic malignant disease.
Cerebrospinal fluid examination revealed lymphocytic pleocytosis with elevated protein and immunoglobin G index, raising concern for an inflammatory process, such as multiple sclerosis. Due to diagnostic uncertainty, the patient was referred to our hospital for further investigation.
On arrival at our hospital, the physical examination demonstrated left-sided hemiparesis. Computed tomography (CT) scans of the chest, abdomen and pelvis did not reveal pathologic findings, with no signs of malignancy.
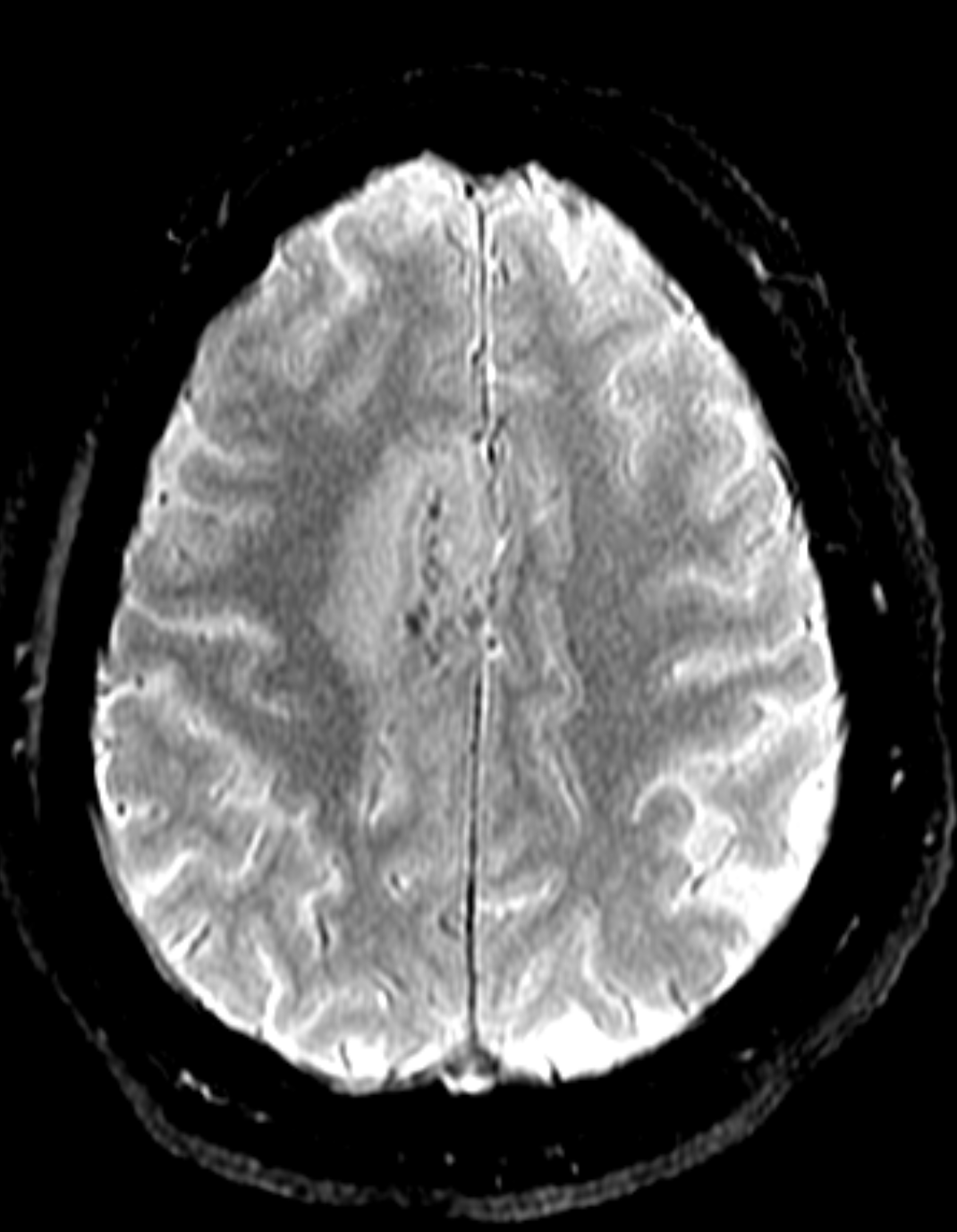
Figure 1C: Blood-sensitive gradient echo (GRE) sequence depicting medial cortical/leptomeningeal susceptibility foci in keeping with cortical superficial siderosis. (Click to enlarge.)
A repeat brain MRI showed a 2.3 cm x 0.9 cm enhancing lesion in the right parasagittal frontal lobe, with extensive leptomeningeal involvement, multiple foci of hemorrhage within the regions of enhancement, and associated vasogenic edema (see Figure 1). MRI spectroscopy showed necrotic tissue, with findings less consistent with glial neoplasm, lymphoma or demyelinating disease (see Figure 2).
Due to inconclusive findings, an open right frontal lobe biopsy was performed. Preliminary results were consistent with necrotizing granulomatous vasculitis (i.e., necrotizing vasculitis including vessels with fibrinoid necrosis in a background of marked inflammation with CD68+ giant cells). No indications of lymphoma, glial neoplasm or microorganisms were found.
Differential diagnoses at that time included primary angiitis of the CNS, isolated CNS sarcoidosis and systemic granulomatous vasculitis.
A review of systems was unremarkable. Inflammatory markers were notable for a mildly elevated C-reactive protein (CPR; 1.4 mg/L) but normal erythrocyte sedimentation rate (ESR; 26 mm/hr), and an extensive autoimmune evaluation was negative. A temporal artery ultrasound was performed, showing no evidence of temporal arteritis. A review of the CT images also did not reveal any signs of systemic vasculitis or sarcoidosis.
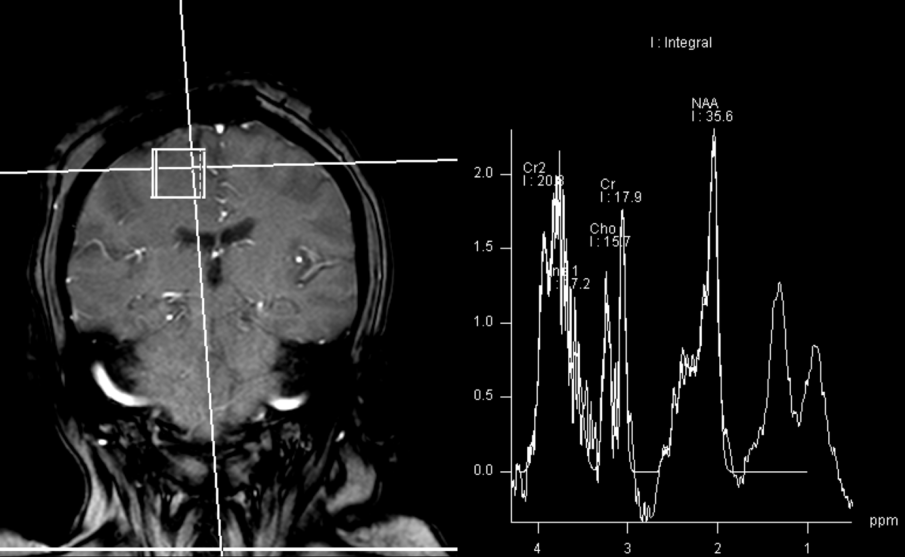
Figure 2: MRI spectroscopy demonstrated decreased NAA (N-acetylaspartate) peak without significant elevation of the choline peak. Findings are suggestive of a necrotic inflammatory process. Spectral features were inconsistent with glial neoplasm or other neoplastic etiologies, such as lymphoma or metastasis. (Click to enlarge.)
Given a low suspicion for infection, empiric treatment was started with pulse-dose glucocorticoids (1 g daily for three days), followed by 1 mg/kg/day of methylprednisolone. The patient exhibited rapid clinical response, with initial improvement in left upper arm strength within days of glucocorticoid initiation. The patient was discharged to a rehabilitation center and received six doses of cyclophosphamide as an outpatient, while remaining on low-dose prednisone (5 mg daily).
The final pathology report showed strong positive staining of the subarachnoid and parenchymal vessels with amyloid β, establishing the diagnosis of inflammatory cerebral amyloid angiopathy (CAA), specifically the cerebral amyloid β-related angiitis (ABRA) subtype (see Figure 3).
At a six-month follow-up in the rheumatology clinic, the patient demonstrated substantial clinical improvement, with complete resolution of her headaches, recovery of left upper extremity strength and improvement in left lower extremity weakness (4/5).
Discussion
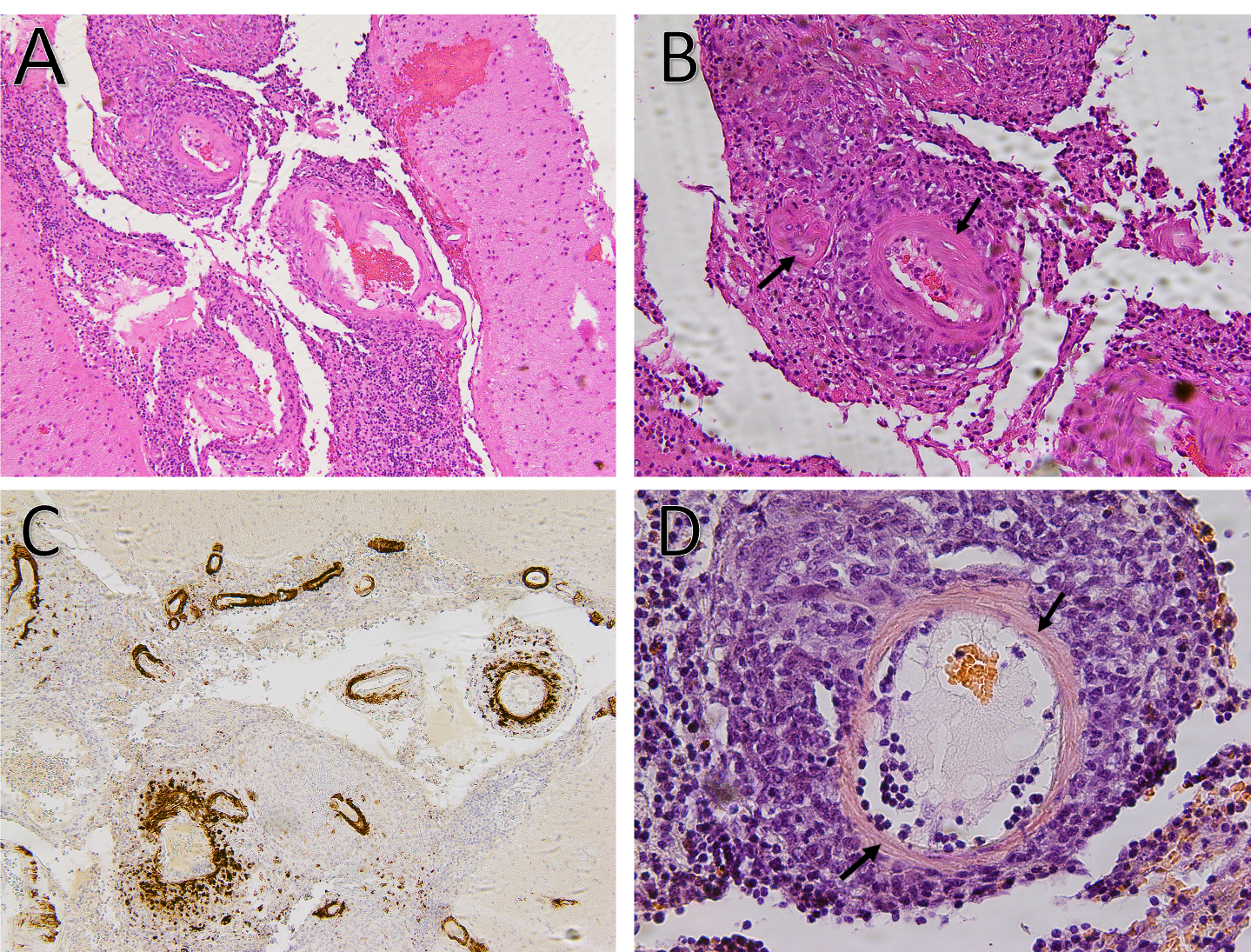
Figure 3A: Hematoxylin and eosin stain demonstrates small to medium-sized vessels that show necrotizing, fibrinoid necrosis representing a necrotizing vasculitis (100x).
Figure 3B: Hematoxylin and eosin stain demonstrates small- to medium-sized vessels that show necrotizing, fibrinoid necrosis representing a necrotizing vasculitis (arrows) (200x).
Figure 3C: Strongly positive amyloid β stain in the wall of subarachnoid blood vessels (100x).
Figure 3D: Salmon-colored tinge of amyloid on the Congo red stain (arrows) (400x). (Click to enlarge.)
Cerebral amyloid angiopathy is a vasculopathy characterized by amyloid β (Aβ) deposition in the cortical and leptomeningeal vessels. It usually manifests in the eighth decade of life, and its clinical features include lobar intracerebral hemorrhage (ICHs), progressive cognitive impairment, transient neurological episodes (TNEs) and epilepsy.6,10-12
Inflammatory CAA is a distinct subset of CAA with specific clinical and neuroradiological features characterized by an inflammatory reaction to the cerebral vascular deposits of amyloid β.7,13 The inflammatory response may be nondestructive and restricted to the lymphocytic perivascular cuffing (CAAri) or may also involve the vessel wall with transmural or intramural, and often granulomatous, inflammatory infiltrates (ABRA). Although pathologically ABRA resembles primary angiitis of the CNS, imaging findings and the colocalization of Aβ deposits, with areas of vasculitic involvement, indicate its relationship with CAAri.5,14
Based on a comprehensive review of ABRA cases by Danve et al., the mean age at the time of diagnosis is 65.2 years old and there is no gender predominance (men 53%).15 Common clinical manifestations include cognitive and behavioral changes (71%), followed by focal neurologic deficits (51%), headaches (35%) and seizures (30%).15
A comparison of ABRA and CAAri cases in a large Chinese population showed no differences in the demographic or clinical characteristics of the two groups.8 On the other hand, patients with ABRA are usually older than patients with primary CNS vasculitis (mean age 45 years) and may have a more acute disease onset.16
Regarding imaging findings, brain T2-weighted MRI sequences, susceptibility-weighted imaging (SWI) and fluid-attenuated inversion recovery (FLAIR) sequences have been widely used to identify patients with suspected inflammatory CAA.17,18 Typical findings include T2/FLAIR hyperintensities in the cortical white matter, usually asymmetric, with or without leptomeningeal involvement, and multiple microhemorrhages.17-20 Susceptibility-weighted imaging sequences and gradient echo (GRE) are abnormal in most patients, revealing cerebral microbleeds and superficial siderosis.6,18 The presence of leptomeningeal involvement, with or without underlying white matter abnormalities, and the absence of lobar hemorrhages help differentiate patients with inflammatory CAA from those with non-inflammatory CAA.14,21
The two pathologic subtypes of inflammatory CAA share many similar imaging findings, but ABRA cases show a higher contrast enhancement (66.7%) compared with CAAri (31.3%), possibly indicating more widespread involvement of vessel walls in ABRA.1 In a small fraction of cases (18%), inflammatory CAA can present as an infiltrative white matter lesion mimicking malignant tumors, as with our patient.5,13,14
The overlapping clinical and imaging features of inflammatory CAA with other inflammatory and non-inflammatory conditions lead to a broad and challenging differential diagnosis.3-6 Histological sampling in the form of stereotactic brain biopsy remains the gold standard for diagnosing inflammatory CAA.
The recognition that typical MRI findings in the appropriate clinical context can indicate the diagnosis of inflammatory CAA prompted the development and validation of diagnostic clinical-radiological criteria. These criteria consider the patient’s age, clinical presentation, white matter hyperintensities and hemorrhagic lesions to classify patients into probable and possible CAAri, avoiding the risks and complications associated with an invasive diagnostic procedure.23 Of note, only patients with the pathologic subtype of CAAri were included in this study. Based on this algorithm, our patient would have been classified with probable CAAri despite the final pathology confirming the presence of the ABRA subtype. This corroborates the notion that the two pathological subtypes of CAA share many clinical and radiological features.
Regarding ancillary tests that can aid in the diagnosis of ABRA, cerebrospinal fluid examination reflects non-specific inflammatory changes, including elevated protein (71%) and lymphocytic pleocytosis (44%).8 Serum inflammatory markers, including CRP and ESR, were elevated in 60% and 37.5% of cases, respectively, in a recent systemic review that included patients with both CAAri and ABRA.24
No systematic evidence evaluating treatment strategies for ABRA or CAAri exists, and most recommendations are borrowed from experience with primary angiitis of the CNS (PACNS). Nevertheless, the effectiveness of immunosuppressive therapy has been confirmed in sizeable patient series in daily practice.25
Chu et al. noticed that 78% of the ABRA patients in their series showed complete or partial response to glucocorticoids.8 High-dose glucocorticoids are typically used, including intravenous 1 g/day methylprednisolone for three to five days, followed by a prolonged tapering dose of oral glucocorticoids.
In the same study and other case series, multiple immunomodulatory agents have been implemented as steroid-sparing agents, including cyclophosphamide, mycophenolate mofetil, methotrexate and azathioprine.8,20,21,24
Caldas et al. noticed that within 21 months, 42% of patients with inflammatory CAA returned to their baseline functional status, 37% died and 20% still had residual functional disabilities following treatment. No difference in outcomes between patients treated with glucocorticoids alone, or in combination with other agents, was noted.24 Interestingly, as demonstrated by Salvarani et al., a subset of patients with prominent leptomeningeal enhancement shows a favorable response to therapy.26
Conclusions & Future Directions
Despite our progress in understanding and managing ABRA, many gaps in knowledge remain. Expanding and validating the proposed clinical-radiological criteria on ABRA cases could further reduce the need for invasive diagnostic tests; identifying subgroups of patients within the spectrum of inflammatory CAA who would need more aggressive management could also be an area of future research; the development of formal treatment guidelines; and consideration of agents used in other granulomatous inflammatory diseases, such as tumor necrosis factor alpha (TNFα) inhibitors could also be included in the future research agenda.
Alexandros Grivas, MD, is a resident in the division of internal medicine at Thomas Jefferson University Hospital, Philadelphia.
Nikil Revuri, MD, is a rheumatologist at Thomas Jefferson University Hospital, Philadelphia. Aleksandra Karaseva, MD, is a resident in the division of pathology at Thomas Jefferson University Hospital, Philadelphia.
Basimah AlBalooshy, MD, is a radiologist at Thomas Jefferson University Hospital, Philadelphia.
Andres M. Ponce, MD, is a rheumatologist at Thomas Jefferson University Hospital, Philadelphia.
References
- Gatti L, Tinelli F, Scelzo E, et al. Understanding the pathophysiology of cerebral amyloid angiopathy. Int J Mol Sci. 2020 May;21(10):3435.
- Chwalisz BK. Cerebral amyloid angiopathy and related inflammatory disorders. J Neurol Sci. 2021 May:424:117425.
- Cho TA, Jones A. CNS vasculopathies: Challenging mimickers of primary angiitis of the central nervous system. Best Pract Res Clin Rheumatol. 2020 Aug;34(4):101569.
- Evangelatos G, Grivas A, Pappa M, et al. Cranial giant cell arteritis mimickers: A masquerade to unveil. Autoimmun Rev. 2022 May;21(5):103083.
- de Souza A, Tasker K. Inflammatory cerebral amyloid angiopathy: A broad clinical spectrum. J Clinical Neurol. 2023 May;19(3):230–241.
- Chwalisz BK. Cerebral amyloid angiopathy and related inflammatory disorders. J Neurol Sci. 2021 May:424:117425.
- Salvarani C, Hunder GG, Morris JM, et al. Aβ-related angiitis: Comparison with CAA without inflammation and primary CNS vasculitis. Neurology. 2013 Oct 29;81(18):1596–1603.
- Chu S, Xu F, Su Y, et al. Cerebral amyloid angiopathy (CAA)-related inflammation: Comparison of inflammatory CAA and amyloid-β-related angiitis. J Alzheimers Dis. 2016;51(2):525–532.
- Auriel E, Charidimou A, Gurol ME, et al. Validation of clinicoradiological criteria for the diagnosis of cerebral amyloid angiopathy-related inflammation. JAMA Neurol. 2016 Feb;73(2):197–202.
- Storti B, Gabriel MM, Sennfält S, et al. Rare forms of cerebral amyloid angiopathy: Pathogenesis, biological and clinical features of CAA-ri and iCAA. Front Neurosci. 2023 Jul:17:1219025.
- Tabaee Damavandi P, Storti B, Fabin N, et al. Epilepsy in cerebral amyloid angiopathy: An observational retrospective study of a large population. Epilepsia. 2023 Feb;64(2):500–510.
- Malhotra K, Theodorou A, Katsanos AH, et al. Prevalence of clinical and neuroimaging markers in cerebral amyloid angiopathy: A systematic review and meta-analysis. Stroke. 2022 Jun;53(6):1944–1953.
- Eng JA, Frosch MP, Choi K, et al. Clinical manifestations of cerebral amyloid angiopathy-related inflammation. Ann Neurol. 2004 Feb;55(2):250–256.
- Salvarani C, Morris JM, Gianni C, et al. Imaging findings of cerebral amyloid angiopathy, aβ-related angiitis (ABRA), and cerebral amyloid angiopathy–related inflammation. Medicine (Baltimore). 2016 May;95(20):e3613.
- Danve A, Grafe M, Deodhar A. Amyloid beta-related angiitis—A case report and comprehensive review of literature of 94 cases. Semin Arthritis Rheum. 2014 Aug;44(1):86–92.
- Salvarani C, Brown Jr RD, Calamia KT, et al. Primary central nervous system vasculitis: Comparison of patients with and without cerebral amyloid angiopathy. Rheumatology (Oxford). 2008 Nov;47(11):1671–1677.
- Corovic A, Kelly S, Markus HS. Cerebral amyloid angiopathy associated with inflammation: A systematic review of clinical and imaging features and outcome. Int J Stroke. 2018 Apr;13(3):257–267.
- Aghetti A, Sène D, Polivka M, et al. Cerebral amyloid angiopathy related inflammation with prominent meningeal involvement. A report of 2 cases. Front Neurol. 2019 Sep:10:984.
- Chung KK, Anderson NE, Hutchinson D, et al. Cerebral amyloid angiopathy related inflammation: Three case reports and a review. J Neurol Neurosurg Psychiatry. 2011 Jan;82(1):20–26.
- Li D, Qin W, Guo Y, et al. Clinical, laboratory, and radiological features of cerebral amyloid angiopathy-related inflammation (CAA-ri): Retrospective, observational experience of a single centre. Neurol Sci. 2023 Feb;44(2):631–638.
- Theodorou A, Palaiodimou L, Malhotra K, et al. Clinical, neuroimaging, and genetic markers in cerebral amyloid angiopathy-related inflammation: A systematic review and metaanalysis. Stroke. 2023 Jan;54(1):178–188.
- Ronsin S, Deiana G, Geraldo AF, et al. Pseudotumoral presentation of cerebral amyloid angiopathy-related inflammation. Neurology. 2016 Mar;86(10):912–919.
- Auriel E, Charidimou A, Gurol ME, et al. Validation of clinicoradiological criteria for the diagnosis of cerebral amyloid angiopathy-related inflammation. JAMA Neurol. 2016 Feb;73(2):197–202.
- Castro Caldas A, Silva C, Albuquerque L, et al. Cerebral amyloid angiopathy associated with inflammation: Report of 3 cases and systematic review. J Stroke Cerebrovasc Dis. 2015 Sep;24(9):2039–2048.
- Regenhardt RW, Thon JM, Das AS, et al. Association between immunosuppressive treatment and outcomes of cerebral amyloid angiopathy-related inflammation. JAMA Neurol. 2020 Oct;77(10):1261–1269.
- Salvarani C, Brown RD, Calamia KT, et al. Primary central nervous system vasculitis with prominent leptomeningeal enhancement: A subset with a benign outcome. Arthritis Rheum. 2008 Feb;58(2):595–603.