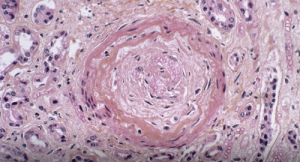
Light micrograph of a vascular lesion (center) caused by systemic sclerosis.
CNRI / Science Source
Systemic sclerosis (SSc) is a multi-system connective tissue disease in which skin and internal organ fibrosis are associated with an obliterative micro-vasculopathy and a degree of inflammation.1 Patients often report it takes one to three years from the appearance of the first signs and symptoms before they receive a diagnosis. The signs and symptoms of systemic sclerosis can be mistaken for any number of diseases, depending on the predominant symptom at the time of examination.3 Early detection provides the opportunity to manage the disease process before damage and fibrosis lead to organ failure and poor outcomes.4
A salt-and-pepper appearance of the skin is just one of many changes observed in the skin of patients with systemic sclerosis.5 It is one of the earliest cutaneous findings in systemic sclerosis.6 Occasionally, it may be the sole cutaneous manifestation.5
Below, we present a case of systemic sclerosis diagnosed five years from the first presentation, which was pseudo-vitiligo, i.e., salt-and-pepper skin pigmentation.
The Case
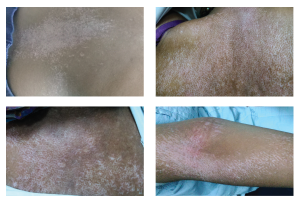
Figures 1, 2, 3 & 4
These photos show salt-and-pepper pigmentation over the patient’s upper back, trunk and upper extremity.
A 30-year-old female patient from Egypt presented to the rheumatology department with chief complaints of pain and swelling of both hands, and thickening and hardening of the skin over her extremities for the past two months. The patient had first become symptomatic in 2013, when her symptoms started as painless skin pigmentation over her neck, back and shoulder, which were diagnosed as vitiligo. At that time, she received systemic psoralen and ultraviolet A therapy for six months with a moderate response.