
Anton Khrupin / Shutterstock.com
A 58-year-old African American woman with a past medical history of hypertension (HTN), hyperlipidemia, severe gastroesophageal reflux disease (GERD) and limited cutaneous systemic sclerosis (lcSSc) presented to the emergency department with shortness of breath (SOB) and progressive bilateral lower extremity swelling for three weeks. She denied any chest pain, but endorsed generalized fatigue and dyspnea with activities of daily living. She denied orthopnea, but stated she always sleeps upright secondary to severe GERD. On review of systems, she endorsed occasional palpitations without presyncopal symptoms and a dry cough. She denied any caffeine intake, stimulant use, alcohol use or illicit drug use.
Her medications included a daily low-dose aspirin, triamterene-hydrochlorothiazide, pravastatin, omeprazole, amlodipine for Raynaud’s symptoms and infrequent use of ibuprofen as needed for pain or headaches.
A CT of her chest, pulmonary embolism protocol, showed bilateral pleural effusions without evidence of parenchymal abnormalities consistent with interstitial lung disease (see Figure 3). An echocardiogram showed a left ventricular ejection fraction of 33%, concentric left ventricular hypertrophy, and an estimated systolic pulmonary artery pressure of 80 mmHg. Cardiac catheterization showed normal coronary arteries, normal pulmonary artery capillary wedge pressure and a pulmonary artery pressure mean of 38 mmHg, indicating mild pulmonary hypertension.
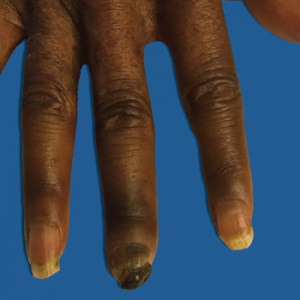
Figure 1: Right Third Digit Necrosis
Patient has mild edema and significant loss of nail, as well as clinical evidence of acroosteolysis of her third digit.
Upon discharge, her diagnoses included hypertensive emergency, acute chronic heart failure with reduced ejection fraction, acute renal failure without evidence of scleroderma renal crisis and mild pulmonary hypertension. Given her dangerously low left ventricular ejection fraction, which can be associated with sudden cardiac death, she was discharged with an external defibrillator vest.
Approximately one month later, she was readmitted to the hospital because of dizziness and light-headedness while sitting in her car, and her ECG on readmission showed a narrow complex tachycardia with non-sinus atrial activity, interpreted as possible atrial tachycardia with atypical atrial flutter. She remained hemodynamically stable, and her symptoms resolved with carvedilol, so she was discharged home after a short hospitalization.
A few weeks later, she was readmitted once again after her external defibrillator vest delivered a shock during continued intermittent palpitations at home, and review of the external defibrillator vest recordings showed supraventricular tachycardia in the 200 bpm range. Repeat ECG was interpreted as atrioventricular nodal re-entrant tachycardia. The electrophysiology team was consulted and performed color mapping, showing areas of scar tissue or fibrosis (see Figure 4). Activation mapping was also done, which revealed the location of an atrial micro-reentry circuit, resulting in atrial flutter (see Figure 5), in the same location as the fibrosis. The electrophysiologist successfully ablated this area, resolving the patient’s recurrent arrhythmias.
Her final discharge diagnosis was recurrent atrial re-entry tachycardia secondary to primary cardiac fibrosis from SSc. At her follow-up visit with a rheumatologist, the patient was feeling well without any symptoms of palpitations, presyncope, SOB or volume overload. She did not require any long-term anti-arrhythmic medication.
Awareness of cardiac manifestation is paramount, because cardiac involvement portends a poor prognosis in SSc.
Discussion
Clinical cardiac manifestations occur in 15–35% of scleroderma (SSc) patients, but the majority of SSc patients have subclinical cardiac involvement at post-mortem autopsy.1,2 Awareness of cardiac manifestation is paramount, because cardiac involvement portends a poor prognosis in SSc.2 Primary cardiac manifestations include fibrosis of the myocardium, pericardium and the conduction system, as well as heart failure and arrhythmia.1 Secondary cardiac manifestations occur secondary to pulmonary hypertension, interstitial lung disease or renal failure resulting in cardio-renal syndrome.1
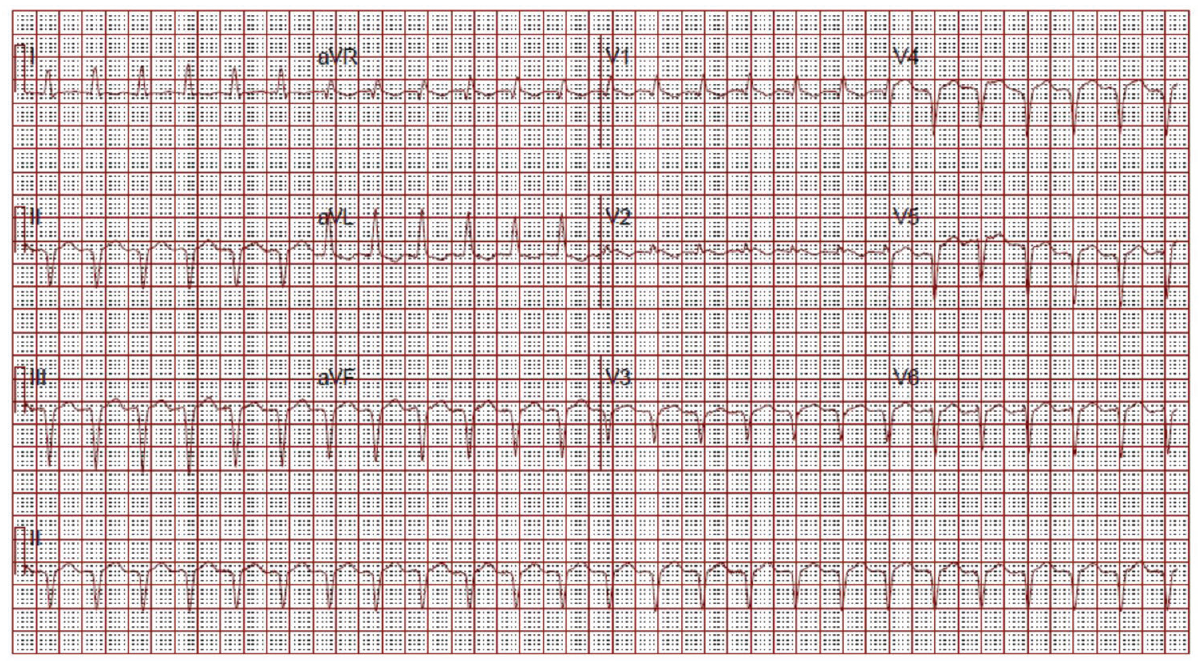
(click for larger image) Figure 2: ECG on Admission
Narrow complex tachycardia. Interpreted as sinus tachycardia.
Primary injury to an organ results in initiation of a fibrogenic response, activation of effector cells, elaboration of extracellular matrix (ECM) and dynamic deposition with insufficient resorption of ECM.3 This is no different when it comes to the heart.
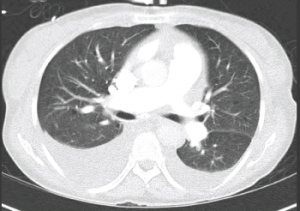
Figure 3: CT Chest
Bilateral pleural effusions. No evidence of pulmonary embolism or cardiomegaly.
As with most organs in the body, the balance of normal architecture is key in normal functioning. Cardiac cells are embedded in a collagen network made up of mostly type I (80%) and type III collagen (11%).4 This collagen network provides physical support, and also sustains transmission of evenly distributed force throughout the myocardium.4 Connexins also play a major role in propagation of electrical impulse through the heart.4 Cardiac fibroblasts are the most abundant cell type in the myocardium.3 These cells are derived from fibroblasts that are native to the myocardium, circulating in the blood or result from epithelial to mesenchymal transition (EMT).3
EMT is a biologic process that allows a polarized epithelial cell, which normally is attached to the basement membrane, to undergo multiple changes that enable it to assume a mesenchymal cell phenotype.5,6 EMT is initiated via inflammation through the activation of transcription factors, expression of specific cell-surface proteins, reorganization and expression of cytoskeletal proteins, production of ECM-degrading enzymes and changes in the expression of specific microRNAs.5,6 The majority of epithelial cells exposed to inflammation undergo apoptosis, but some transition and have enhanced migratory capacity, invasiveness, elevated resistance to apoptosis.5,6 These cells that have undergone EMT have greatly increased production of ECM components after transition.
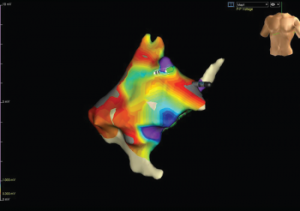
Figure 4: Color Mapping of the Right Atrium
Color mapping is done by electrophysiology to show the amplitude of voltage to evaluate for areas of scarring or fibrosis. Grey areas indicate areas of fibrosis.
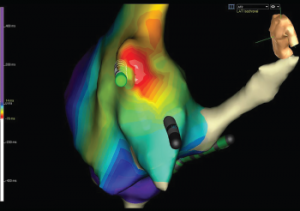
Figure 5: Activation Mapping of the Right Atrium
Activation mapping is done by electrophysiology to evaluate the speed of conduction in the heart. White represents areas of early conduction, which then radiates to slower conduction via change in colors from the spectrum starting with red. The white area is the patient’s re-entrant nidus, and this is the location that the electrophysiologist ablated, resolving the recurrent arrhythmias.
All of these factors, when out of balance with increased deposition of ECM with decreased resorption, result in myocardial fibrosis.5,6 As with the pathophysiology of fibrosis throughout all body tissues in SSc patients, TGF-β via the Smad pathway is one of the key players in cardiac fibrosis as well.3,7
Cardiac complications vary on the basis of the pattern of cardiac fibrosis.4 Patchy fibrosis, in which collagen fibers and myocardial bundles intermingle, is the most proarrhythmic pattern of fibrosis. On the other hand, compact fibrosis is the least proarrhythmic, so when electrophysiologists ablate areas of arrhythmia origination, the goal is to create compact fibrosis to prevent abnormal signal propagation. This is the technique that was successfully completed in our patient’s case.
Conclusion
Understanding the risks of cardiac complications in SSc patients is important for the rheumatologist because cardiac involvement results in increased morbidity and mortality in this patient population.8 In fact, a mere 3% increase in extracellular volume fraction of fibrous tissue in the myocardium is associated with a 50% increase in the risk of adverse cardiac events.9 Keeping in mind that cardiac complications can be subclinical, it is important to risk stratify SSc patients for appropriate screening.
In conclusion, the next time an SSc patient reports palpitations during a review of systems, remember that it could be more than just a feeling.
 Sophia C. Weinmann, MD, recently completed a rheumatology fellowship at Washington University in St. Louis. She is now a medical instructor at Duke University, Durham, N.C.
Sophia C. Weinmann, MD, recently completed a rheumatology fellowship at Washington University in St. Louis. She is now a medical instructor at Duke University, Durham, N.C.
 Richard D. Brasington Jr., MD, FACP, is a professor of medicine and the rheumatology fellowship program director at Washington University in St. Louis. He is the recipient of multiple teaching awards that recognize his dedication to rheumatology.
Richard D. Brasington Jr., MD, FACP, is a professor of medicine and the rheumatology fellowship program director at Washington University in St. Louis. He is the recipient of multiple teaching awards that recognize his dedication to rheumatology.
References
- Lambova S. Cardiac manifestations in systemic sclerosis. World J Cardiol. 2014 Sep 26;6(9):993–1005.
- Kahan A, Coghlan G, McLaughlin V. Cardiac complications of systemic sclerosis. Rheumatology (Oxford). 2009 Jun;48(Suppl 3):
- 45–48.
- Rockey DC, Bell PD, Hill JA. Fibrosis—A common pathway to organ injury and failure. N Engl J Med. 2015 Mar 19;372(12):1138–1149.
- De Jong S, Van Veen TAB, Van Rijen HVM, De Bakker JMT. Fibrosis and cardiac arrhythmias. J Cardiovasc Pharmacol. 2011 Jun;57(6):630–638.
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009 Jun;119(6):1420–1428.
- Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003 Dec;112(12):1776–1784.
- Van Rooij E, Olson EN. Searching for MiR‑acles in cardiac fibrosis. Circ Res. 2009 Jan 30;104(2):138–140.
- Champion HC. The heart in scleroderma. Rheum Dis Clin North Am. 2008 Feb; 34(1):181–190.
- Wong TC, Piehler K, Meier CG, et al. Association between extracellular matrix expansion quantified by cardiovascular magnetic resonance and short-term mortality. Circulation. 2012 Sep 4;126(10):1206–1216.
