 Pachydermoperiostosis (PDP), also known as Touraine-Solente-Golé syndrome or primary hypertrophic osteoarthropathy, is a rare syndrome that can be inherited as autosomal dominant, autosomal recessive, or sporadically. This progressive disease primarily affects males, who tend to have more severe features than females. PDP usually occurs during adolescence, often starting around puberty.1 The main clinical features are clubbing, pachydermia and hyperhidrosis. Fortunately, it is a self-limiting disease. However, it can be difficult to diagnose and treat. This report describes the current known pathogenesis of PDP and possible associated abnormalities that could explain potential gastrointestinal (GI) manifestations.
Pachydermoperiostosis (PDP), also known as Touraine-Solente-Golé syndrome or primary hypertrophic osteoarthropathy, is a rare syndrome that can be inherited as autosomal dominant, autosomal recessive, or sporadically. This progressive disease primarily affects males, who tend to have more severe features than females. PDP usually occurs during adolescence, often starting around puberty.1 The main clinical features are clubbing, pachydermia and hyperhidrosis. Fortunately, it is a self-limiting disease. However, it can be difficult to diagnose and treat. This report describes the current known pathogenesis of PDP and possible associated abnormalities that could explain potential gastrointestinal (GI) manifestations.
Case Presentation
A 16-year-old young man presented with a history of protein-losing enteropathy, recurrent diarrhea secondary to Clostridioides difficile (C. diff) infections, iron deficiency and concern for eosinophilic esophagitis or Crohn’s disease. He had previously been healthy, with no pertinent family history, until he developed a gagging sensation at about 13 years old.
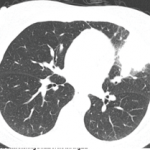
Double-balloon enteroscopy with a capsule endoscopy was performed, which noted two proximal ileal strictures. A biopsy of the ulcerated stricture area showed mild, active ileitis with mixed inflammatory infiltrate, focal ulceration and reactive changes.
Repeat esophagogastroduodenoscopy/colonoscopy showed mild inflammation of the distal colon and lymphoplasmacytosis. Fecal calprotectin fluctuated between abnormal, borderline and normal. Inflammatory markers were mildly abnormal, with the erythrocyte sedimentation rate (ESR) fluctuating between 29 and 53 mm/hr (reference range [RR]: <11 mm/hr) and C-reactive protein (CRP) between 1.0 and 4.7 mg/dL (RR: <1 mg/dL).
In the past, the patient had tested positive multiple times for stool α1-antitrypsin, which helped lead to a diagnosis of protein-losing enteropathy. He was initially treated with metronidazole, vancomycin and fidaxomicin due to multiple C. diff infections.
Figure 1
One-and-a-half years prior to this presentation, he had complained of knee and foot swelling. The pain and swelling worsened with activity, and he had daily morning stiffness for 10 minutes. He was evaluated by a cardiologist for foot edema, but had a normal echocardiogram. Ultrasound of his extremities ruled out deep vein thromboses. His symptoms progressed, and he developed hand swelling, with limited range of motion. Due to worsening symptoms, he was admitted for further evaluation and a rheumatologist was consulted.