MOLEKUUL / Science Source
Drug-induced lupus erythematosus and ANCA-associated vasculitis (AAV) are both autoimmune conditions associated with the use of hydralazine, a commonly prescribed drug for hypertension and congestive heart failure. Although the pathogenesis is unknown, it is believed that hydralazine alters neutrophil and lymphocyte function and promotes exposure of antigens, leading to the development of anti-neutrophil antibodies (ANCA) and/or anti-nuclear antibodies (ANA) and ultimately triggering a systemic response.1
These conditions are rare and typically considered to be mutually exclusive. Here, we describe a rare presentation in which hydralazine appears to be the inciting factor of a patient developing an overlap of drug-induced lupus erythematosus and AAV.
During her hospitalization, infection was ruled out and ocular findings were determined to be consistent with episcleritis.
Laboratory data revealed proteinuria and hematuria on initial urinalysis. Serum creatinine was elevated from her normal baseline at 1.44 mg/dL (normal: 0.50–1.40 mg/dL) with a BUN (blood urea nitrogen) level of 39 mg/dL (normal: 7–22 mg/dL). Her urine creatinine was 86.8 mg/dL and urine protein was 335 mg/dL, with a ratio of 3.86.
The complete blood count revealed leukopenia 3.6 cells/µl (normal: 4.5–10 cells/µl), and lymphopenia 10.1% (normal: 20–40%). An autoimmune evaluation was remarkable for ANA (1:640 speckled), anti-double-stranded DNA antibodies (1:10), myeloperoxidase (MPO) ANCA, and P-ANCA 1:320. Her C3 level was 69 mg/dL (normal: 88–201 mg/dL) and C4 was 10 mg/dL (normal: 16–44 mg/dL). Her C-reactive protein was elevated at 214 mg/L (<2.9 mg/L), as was the sedimentation rate at >100 mm/Hr (normal: 0–20 mm/hr). Her anti-histone immunoglobulin (Ig) G was 5.0 (normal: 0.0–0.9 units). Anti-Smith, anti-RNP and anti-SSA/SSB antibodies were negative. Antiphospholipid antibodies were negative. Cryoglobulin, anti-glomerular basement membrane antibodies and proteinase-3 (PR3) ANCA were undetectable.
CT imaging revealed stenosis at the left subclavian artery, multiple sub-centimeter pulmonary nodules, cardiomegaly and trace pericardial and pleural effusions. Additionally, incidental findings of hepatic and renal cysts were noted.
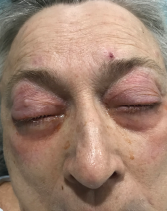
Figure 1A
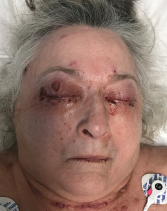
Figure 1B
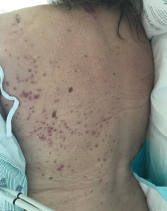
Figure 1C
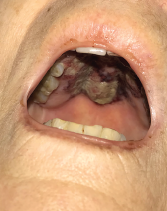
Figure 1D
The patient presented with periorbital edema, erythematous conjunctiva, papular lesions all over her body and a mucosal ulceration on the hard palate of her oral cavity.
A biopsy of the buccal mucosa revealed stratified squamous epithelium and underlying subepithelial connective tissue with marked acute and chronic inflammation, ulceration and necrosis.
Stains for cytomegalovirus and herpes simplex virus were negative, and GMS and gram stains were negative for fungal elements and pathogenic bacteria. Mild perivascular inflammation was noted, but overt vasculitis was not seen. Features of Stevens-Johnson syndrome were also absent.
The renal biopsy revealed diffuse mesangial matrix expansion consistent with diabetic glomerulosclerosis class IIb (without nodular glomerulosclerosis) and very focal acute segmental necrotizing glomerulonephritis affecting 1 of 70 glomeruli in paraffin sections.
Immunofluorescence stains were negative for IgG, IgA, kappa light chain, lambda light chain, C3 and C1q. Weak mesangial deposition of IgM was noted. Mild tubular epithelial injury was present, and there were focal red blood cell casts. Approximately 12% of glomeruli were global sclerotic (12/99 glomeruli in the specimen), and there was 10% interstitial fibrosis with tubular atrophy.
Overall, the renal biopsy was consistent with ANCA-associated vasculitis given the presence of MPO-ANCA and the findings of pauci-immune complex deposition. However, the presence of serum anti-histone antibodies was consistent with drug-induced lupus.
The patient was treated with 1,000 mg of intravenous methylprednisolone daily for five days, then switched to 60 mg of prednisone daily and tapered thereafter. With this treatment, her hematuria and proteinuria considerably improved, and her C3 and C4 levels normalized.
Although hydralazine was discontinued, the patient’s condition remained active on a tapered dose of steroids. At that time, the patient was started on 375 mg/m2 rituximab weekly for four doses. Following this additional immunosuppression, her renal function normalized and her anemia resolved.
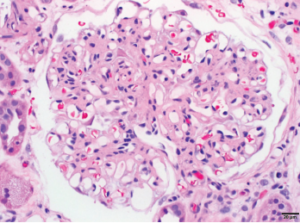
1A: Glomerulus with moderate global mesangial matrix expansion consistent with moderately advanced diabetic nephropathy (without nodular glomerulosclerosis) is noted.
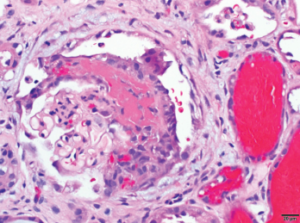
1B: Focal glomerulus with segmental fibrinoid necrosis with extracapillary proliferation and red blood cells casts consistent with necrotizing, pauci-immune, ANCA-associated glomerulonephritis are noted. Bar=20 μm.
Conclusion
Hydralazine is a common medication for congestive heart failure and hypertension that has been associated with systemic autoimmune conditions including drug-induced lupus, ANCA-associated vasculitis and leukocytoclastic vasculitis. It is thought that hydralazine accumulates in the intracytoplasmic neutrophilic granules, which in turn, leads to binding of myeloperoxidase and ultimately causes release of cytotoxic products and cell death. Once these neutrophils undergo cell death, antigens that are normally sequestered are exposed. This exposure leads to uptake by antigen-presenting cells, leading to production of ANCA.2,3
In mild cases of drug-induced autoinflammatory syndrome, cessation of the inciting agent is adequate to resolve further morbidity.
Historically, AAV and drug-induced lupus erythematosus have been considered mutually exclusive, but our case highlights an overlap, suggesting the possibility of a continuum between these conditions.
Additionally, hydralazine-associated AAV in the literature suggests a predominance of renal-limited vasculitis. Our case, however, describes pulmonary-renal syndrome.4,5
In mild cases of drug-induced autoimmune syndromes, cessation of the inciting agent is adequate to resolve further morbidity. However, our case suggests that drug cessation may not be enough to address the phenomenon of vasculitis and further immunosuppression may be warranted.
 Mohammad A. Ursani, MD, FACP, RhMSUS, is a private practice rheumatologist in The Woodlands, Texas, and serves as an adjunct faculty member for the University of Texas, Houston, Internal Medicine Residency Program. He also serves as a volunteer for the ACR and is a young physician ambassador for the Harris County Medical Society.
Mohammad A. Ursani, MD, FACP, RhMSUS, is a private practice rheumatologist in The Woodlands, Texas, and serves as an adjunct faculty member for the University of Texas, Houston, Internal Medicine Residency Program. He also serves as a volunteer for the ACR and is a young physician ambassador for the Harris County Medical Society.
 Ojas Naik, MD, is a private practice nephrologist at Kidney Specialists of North Houston with an interest in autoimmune manifestations of renal disease.
Ojas Naik, MD, is a private practice nephrologist at Kidney Specialists of North Houston with an interest in autoimmune manifestations of renal disease.
 Rohaan Khan is a health sciences student at Michael E. DeBakey High School for Health Professions and has an interest in pursuing a degree in medicine.
Rohaan Khan is a health sciences student at Michael E. DeBakey High School for Health Professions and has an interest in pursuing a degree in medicine.
 William F. Glass II, MD, PhD, is a professor and vice chair for graduate medical education, director of the pathology residency program and director of renal pathology services at the University of Texas, Houston.
William F. Glass II, MD, PhD, is a professor and vice chair for graduate medical education, director of the pathology residency program and director of renal pathology services at the University of Texas, Houston.
References
- Grau RG. Drug-induced vasculitis: New insights and a changing lineup of suspects. Curr Rheumatol Rep. 2015 Dec;17(12):71.
- Hogan JJ, Markowitz GS, Radhakrishnan J. Drug-induced glomerular disease: Immune-mediated injury. Clin J Am Soc Nephrol. 2015 Jul 7;10(7):1300–1310.
- Agarwal G, Sultan G, Werner SL, Hura C. Hydralazine induces myeloperoxidase and proteinase 3 anti-neutrophil cytoplasmic antibody vasculitis and leads to pulmonary renal syndrome. Case Rep Nephrol. 2014;2014:868590.
- Yokogawa N, Vivino FB. Hydralazine-induced autoimmune disease: Comparison to idiopathic lupus and ANCA-positive vasculitis. Mod Rheumatol. 2009;19(3):338–347.
- Suneja M, Baiswar S, Vogelgesang SA. Hydralazine associated pauci-immune glomerulonephritis. J Clin Rheumatol. 2014 Mar;20(2):99–102.