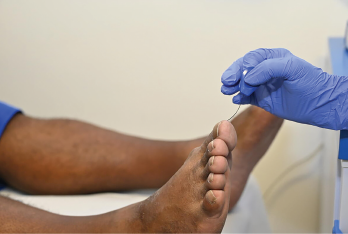
A monofilament test is being carried out on the foot of a 50-year-old male patient to test for nerve damage (neuropathy).
DR P. MARAZZI/Science Source / shutterstock.com
Granulomatosis with polyangiitis (GPA), is a type of anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitis (AAV) that affects small- to medium-sized vessels.1 It can occur equally in both men and women, with a reported mean age at onset of 55 years.2
Here, we present the case of a 59-year-old man with GPA who initially presented with vasculitic neuropathy and later developed renal involvement.
Case Presentation
A 59-year-old man with a past medical history significant for GPA came to our rheumatology clinic to establish care.
His symptoms started with recurrent episodes of sinusitis that led to multiple otolaryngologist visits, several courses of antibiotics and, eventually, placement of tympanostomy tubes. He then developed acute onset, progressive numbness and weakness of both legs, which resulted in the patient becoming wheelchair bound.
A nerve conduction study was notable for the low amplitude compound muscle action potentials. This finding prompted the patient to undergo a left sural nerve biopsy, which showed evidence of vasculitis of the epineural vessels. Laboratory studies revealed elevated markers of inflammation, positive PR3-cANCA/c-ANCA proteinase 3 anti-neutrophil cytoplasmatic antibodies, negative anti-nuclear antibody (ANA), anti-SS-A antibody, anti-SS-B antibody, anti-Scl-70 antibody, anti-double-stranded DNA antibody (anti-dsDNA) and cryoglobulins. Left ankle magnetic resonance imaging showed plantar fasciitis. A computerized tomography scan of his chest was within normal limits. At that time, the patient did not have any other organ involvement, including the lungs and kidneys.
The patient was started on high-dose steroids and 375 mg/m2 of rituximab for four weeks, along with five rounds of plasmapheresis. He noticed a slow, but sustained, improvement over one year. His muscle strength also improved, and he was able to walk. Afterward, he was gradually weaned off prednisone and maintained on 2 g of mycophenolate mofetil (MMF) daily.
Following the improvement in strength, the patient self-discontinued MMF and remained asymptomatic. He was referred to our rheumatology clinic for a second opinion when laboratory testing showed elevated creatinine, microscopic hematuria and an elevated protein-to-creatinine ratio a few years after his initial diagnosis.
At the time of his visit to our clinic, the patient’s laboratory studies revealed a white blood cell count of 11,200 uL (reference range [RR]: 3,800–10,800 uL), platelets of 406,000 uL (RR: 140,000–400,000 per uL), C-reactive protein (CRP) of 48.3 mg/L (RR: <8 mg/L), C-ANCA titer of 1:80 (RR: <1:20 titer), PR3-ANCA 5.5 (RR: <1 antibody index) and a serum creatinine of 1.9 mg/dL (RR: 0.7–1.33 mg/dL). The urinalysis showed 3+ protein (RR: negative), RBC 3–10 (RR: ≤2 per HPF), no hyaline cast or squamous epithelial cells. The urine protein-to-creatinine ratio was 754 mg/g (RR: 22–128 mg/g). On examination, his muscle strength was 5/5 in both his arms and legs. No skin rashes were noted. An examination of his respiratory system was unremarkable. Other systems were also within normal limits. Computed tomography (CT) scans of the chest and sinuses were unremarkable.
The patient refused to undergo a kidney biopsy and was restarted on high-dose steroids for presumptive glomerulonephritis from his GPA. He received 375 mg/m2 of rituximab weekly for four weeks with improvement in his creatinine to 1.28 mg/dL (RR: 0.7–1.33 mg/dL), normalization of CRP (RR: <8 mg/dL), negative C-ANCA (RR: <1:20 titer) and PR3-ANCA (RR: <1 antibody index). Urinalysis showed persistent 3+ protein (normal: negative) and urine protein to creatinine ratio of 242 mg/g creatinine (RR: 22–128 mg/g creatinine), which was an improvement. The hematuria improved following treatment to 0 RBC (RR: ≤2 per HPF). Currently, he is receiving 500 mg of intravenous rituximab every six months and 5 mg of prednisone daily. He does not have any residual weakness, and his muscle strength in all of his extremities is normal.
Discussion
AAV is a group of rare diseases with characteristics of inflammatory cell infiltration and blood vessel wall necrosis. It commonly affects small and medium-size arteries. AAV syndromes include eosinophilic granulomatosis with polyangiitis (EGPA), microscopic polyangiitis (MPA) and GPA.
GPA predominantly involves capillaries, venules, arterioles, arteries and veins. The most common initial presentation of GPA is upper airway disease. Involvement of ears, nose and throat has been reported to be as high as 90% in patients with GPA.2 Other clinical manifestations, such as cough, dyspnea, stridor, wheezing and hemoptysis, have been observed in these patient populations, but are mainly seen in the setting of airway or pulmonary parenchyma involvement.6
Renal involvement is commonly reported in GPA and other forms of AAV. Studies have shown that evidence of glomerulonephritis can be found in only 18% of patients at presentation, but 77–85% of patients will eventually develop glomerulonephritis further along their disease course.2,7
Other manifestations of GPA include skin lesions, such as leukocytoclastic angiitis, urticaria, livedo reticularis and nodules. Orbital manifestations include uveitis, conjunctivitis, episcleritis, scleritis, corneal ulceration, optic neuropathy and retinal vasculitis.8
Vasculitic neuropathy is commonly associated with musculoskeletal, skin & cardiovascular manifestations.
Patients with GPA can also have clinical symptoms of peripheral and central nervous system involvement. Peripheral neuropathy is the most common disorder in this population. Central nervous system involvement is rare. According to a study of 128 patients with GPA by De Groot et al., almost half of the patients developed neurologic symptoms. Of those, 43.8% had peripheral nervous system involvement in the form of sensorimotor polyneuropathy or mononeuritis multiplex, and only 7% had central nervous system involvement.9
Overall, neurologic involvement is uncommon at presentation of GPA. We observed the same finding in our patient, making this case an unusual presentation of GPA. Ripellino et al. reported two cases of peroneal nerve involvement as an initial manifestation of small and medium-size vasculitis that made diagnosis challenging.10
Insights from the Diagnostic and Classification Criteria in Vasculitis (DCVAS) study by Bischof et al. reveal vasculitic neuropathy may not have an association with multi-organ involvement, although developing neuropathy serves as a poor prognostic factor in patients with GPA.5,11 Bischof et al. also found an increased rate of musculoskeletal involvement (e.g., myalgias and weakness), as we observed in our patient.
Conclusion
Although they are more common in advanced stages, the neurologic manifestations of GPA can occur at any time during the disease. Peripheral neuropathy is the most common disorder. However, symmetrical, distal motor-sensory neuropathy, cranial nerve involvement, external ophthalmoplegia and cerebral infarction have been reported.
A high index of suspicion and early biopsy are needed for the timely initiation of immune suppressants. Other systemic manifestations can occur at any point of the disease. These patients should be closely monitored with an extensive review of systems, clinical exams and serology workup during each follow-up visit.
 Ann George, MD, completed a rheumatology fellowship at the University of Central Florida, HCA Healthcare, Orlando. She is now a rheumatologist at Tristate Arthritis Group, Crestview Hills, Ky.
Ann George, MD, completed a rheumatology fellowship at the University of Central Florida, HCA Healthcare, Orlando. She is now a rheumatologist at Tristate Arthritis Group, Crestview Hills, Ky.
 Azin Azarfar, MD, completed an internal medicine residency at the University of Central Florida, HCA Healthcare, Orlando. She is now a first-year rheumatology fellow at the University of Florida, Gainesville.
Azin Azarfar, MD, completed an internal medicine residency at the University of Central Florida, HCA Healthcare, Orlando. She is now a first-year rheumatology fellow at the University of Florida, Gainesville.
 Shazia Bég, MD, is the associate program director of the Rheumatology Fellowship Program and an associate professor of rheumatology in the graduate medical education program at the University of Central Florida, HCA Healthcare, Orlando.
Shazia Bég, MD, is the associate program director of the Rheumatology Fellowship Program and an associate professor of rheumatology in the graduate medical education program at the University of Central Florida, HCA Healthcare, Orlando.
 Maria Farooq, MD, is an assistant professor of rheumatology in the graduate medical education program at the University of Central Florida, HCA Healthcare, Orlando.
Maria Farooq, MD, is an assistant professor of rheumatology in the graduate medical education program at the University of Central Florida, HCA Healthcare, Orlando.
Disclosures
This research was supported (in whole or in part) by HCA Healthcare and/or an HCA Healthcare-affiliated entity. The views expressed in this publication represent those of the author(s) and do not necessarily represent the official position of HCA Healthcare.
References
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013 Jan;65(1):1–11.
- Seo P, Stone JH. The antineutrophil cytoplasmic antibody-associated vasculitides. Am J Med. 2004 Jul;117(1):39–50.
- Almouhawis HA, Leao JC, Fedele S, et al. Wegener’s granulomatosis: A review of clinical features and an update in diagnosis and treatment. J Oral Pathol Med. 2013 Aug;42(7):507–516.
- Nishino H, Rubino FA, DeRemee RA, et al. Neurological involvement in Wegener’s granulomatosis: an analysis of 324 consecutive patients at the Mayo Clinic. Ann Neurol. 1993 Jan;33(1):4–9.
- Bischof A, Jaeger VK, Hadden RD, et al. Peripheral neuropathy in antineutrophil cytoplasmic antibody-associated vasculitides: Insights from the DCVAS study. Neurol Neuroimmunol Neuroinflamm. 2019 Sep 20;6(6):e615.
- Gómez-Puerta JA, Hernández-Rodríguez J, López-Soto A, et al. Antineutrophil cytoplasmic antibody-associated vasculitides and respiratory disease. Chest. 2009 Oct;136(4):1101–1111.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: An analysis of 158 patients. Ann Intern Med. 1992 Mar 15;116(6):488–498.
- Fauci AS, Haynes BF, Katz P, et al. Wegener’s granulomatosis: prospective clinical and therapeutic experience with 85 patients for 21 years. Ann Intern Med. 1983 Jan;98(1):76–85.
- De Groot K, Schmidt DK, Arlt AC, et al. Standardized neurologic evaluations of 128 patient with Wegener’s granulomatosis. Arch Neurol. 2001 Aug;58(8):1215–1221.
- Ripellino P, Varrasi C, Maldi E, et al. Peroneal nerve involvement as initial manifestation of primary systemic vasculitis. BMJ Case Rep. 2014 Mar 31;2014:bcr2014204084.
- Samson M, Puéchal X, Devilliers H, et al. Mononeuritis multiplex predicts the need for immunosuppressive or immunomodulatory drugs for EGPA, PAN and MPA patients without poor-prognosis factors. Autoimmun Rev. 2014 Sep;13(9):945–953.