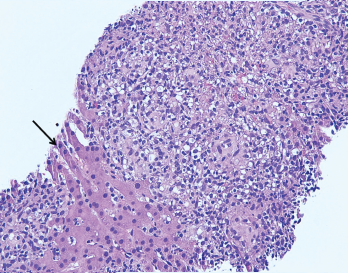
Figure 2. The image from Figure 1 at 200X showing granulomas accompanied by scattered admixed lymphocytes without discernible atypia. Adjacent hepatocytes (black arrow), polygonal cells with centrally placed nucleus and eosinophilic cytoplasm, can also be identified.
A liver biopsy performed on hospital day 3 revealed granulomatous hepatitis (see Figures 1 & 2) but no hemophagocytosis. Iron stains, periodic acid-Schiff diastase stain, immunohistochemistry for cytomegalovirus, and in situ hybridization for Epstein-Barr virus RNA were negative. Subsequent Grocott’s methenamine silver (GMS) stain showed scattered yeast forms (see Figure 3), morphologically consistent with Histoplasma capsulatum. Acid-fast bacillus and Gram stains were negative. Liver tissue culture was not obtained. Serum and urine histoplasma antigens and 1,3 beta-D-glucan were elevated.
A repeat CT of her chest with contrast demonstrated left lower lobe consolidation and incidentally revealed multiple hypodense splenic lesions attributed to a suspected underlying infectious process.