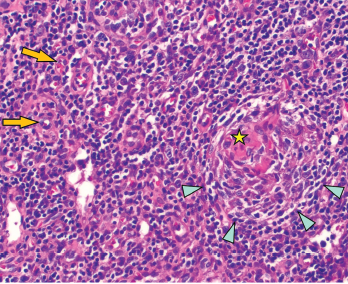
Figure 2: Regressed germinal center with follicular dendritic cell prominence (star), onion-skinning (blue arrowhead) and increased vascularity (orange arrows) are seen.
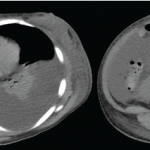
On exam, the patient was febrile, hypotensive and tachycardic. She did not have any eye inflammation, oral or nasal ulcers, abnormal cardiac or pulmonary exam, skin rash or synovitis. There was subcentimeter axillary lymphadenopathy, but no cervical or inguinal lymphadenopathy. A neurologic exam was limited due to sedation, but rightward gaze deviation was noted initially on arrival.
Laboratory values showed pancytopenia, with a white blood cell count of 1.0 x109/L (RR: 3.2–9.8 x109/L), hemoglobin 7 g/dL, platelet 10×109/L (RR: 3.2–9.8×109/L), creatinine 2 mg/dL (RR: 0.4–1.0 mg/dL), AST 200 U/mL (RR: 15–40 U/L), ALT 600 U/mL (RR: 14–50 U/L), ESR 30 mm/hour, CRP 8 mg/dL, ferritin >15,000 ng/mL, triglycerides 500 mg/dL (RR: <500 mg/dL), lactate dehydrogenase 4,000 U/L (RR: 100–200 U/L), sIL-2R 8,000 U/mL (RR: <1,100 U/mL), and absent NK cell activity. CSF studies were normal.