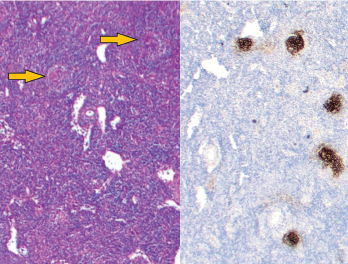
Figure 1: Vague nodular areas (orange arrows) on the left and CD21 staining for follicular dendritic cells on the right demonstrate follicles with regressed germinal centers.
The difference between Castleman disease and Castleman-like disease may be subtle, but it comes with significant ramifications.
Case Presentation
This case involves a pregnant 19-year-old woman who presents over multiple hospitalizations with concerns for systemic lupus erythematosus and macrophage activation syndrome.
She was admitted for further examination and testing, at which time she was noted to have elevated inflammatory markers with an erythrocyte sedimentation rate (ESR) of 65 mm/hour (RR: 0–15 mm/hr) and a C-reactive protein (CRP) of 2.8 mg/dL (RR: <0.60 mg/dL). Her ANA was 1:2560 titer in a homogeneous pattern, and complement levels were low: C3: 74 mg/dL (RR: 86–208 mg/dL); and C4: 12 mg/dL (RR: 13–47 mg/dL). Anti-Ro, anti-La, anti-Smith, anti-RNP, anti-dsDNA, anti-smooth muscle and anti-mitochondrial antibodies were negative.
Due to her significantly elevated bile acids, she was given betamethasone intramuscularly to accelerate fetal lung maturity, and underwent emergent Cesarean section. Her symptoms and laboratory values quickly improved, and she was discharged.
Four months later, she presented to the emergency department with abdominal pain, joint pain and a temperature of 103°F, at which time she was noted to have altered mentation with slow, circular speech. Her laboratory values were significant for hemoglobin of 7 g/dL (RR: 12–15 g/dL), haptoglobin 4 mg/dL (RR: 30–200 mg/dL), direct Coombs’ with IgG 3+ (normal=negative), normal creatinine, urinalysis with trace blood and a 24-hour urine protein of 2,200 mg/g (RR: <100 mg/g). Her inflammatory markers were markedly elevated, with ESR of 117 mm/hour (RR: 0–15 mm/hr), CRP 9.8 mg/dL (RR: <0.60 mg/dL) and ferritin greater than 15,000 ng/mL (RR: 10–200 ng/mL).
Cerebral spinal fluid (CSF) studies were noninflammatory with two nucleated cells x106/L (RR: 0–5×106/L), protein 11 mg/dL (RR: 15–50 mg/dL) and glucose 76 mg/dL. There were no CSF oligoclonal bands (normal: <4 bands) and infectious studies were negative.
Given the patient’s hyperferritinemia, there was concern for macrophage activation syndrome secondary to systemic lupus erythematosus (SLE).
She was treated with dexamethasone and subcutaneous anakinra with rapid improvement of abdominal pain, joint pain and mental status. Despite being drawn two days after immunosuppression was initiated, the soluble interleukin 2 receptor (sIL-2R) was elevated at 2,200 U/mL (RR: <1,100 U/mL. NK cell function was normal.
Given her degree of proteinuria, a renal biopsy was performed. It showed podocyte hypertrophy and effacement, without evidence of immune-complex-mediated disease, focal segmental glomerulosclerosis or thrombotic microangiopathy. Given the possibility of atypical renal involvement from SLE, she was prescribed mycophenolate mofetil and prednisone (of note, patient’s baby was formula fed and not breastfed). Unfortunately, the patient did not follow up as an outpatient, and it was unclear whether she took the prescribed medications.
Two months later, the patient was found unresponsive at home after a generalized tonic-clonic seizure and brought by paramedics to the emergency department, with a temperature of 106°F, hypotensive and tachycardic. She was intubated, placed on a norepinephrine infusion and started on broad-spectrum empiric antibiotics. Other than the past medical history of SLE from recent months, she did not have known comorbidities.
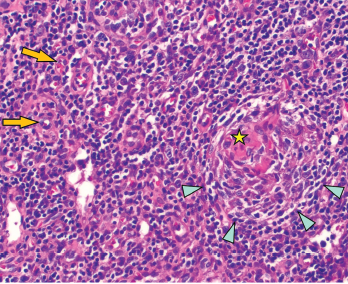
Figure 2: Regressed germinal center with follicular dendritic cell prominence (star), onion-skinning (blue arrowhead) and increased vascularity (orange arrows) are seen.
On exam, the patient was febrile, hypotensive and tachycardic. She did not have any eye inflammation, oral or nasal ulcers, abnormal cardiac or pulmonary exam, skin rash or synovitis. There was subcentimeter axillary lymphadenopathy, but no cervical or inguinal lymphadenopathy. A neurologic exam was limited due to sedation, but rightward gaze deviation was noted initially on arrival.
Laboratory values showed pancytopenia, with a white blood cell count of 1.0 x109/L (RR: 3.2–9.8 x109/L), hemoglobin 7 g/dL, platelet 10×109/L (RR: 3.2–9.8×109/L), creatinine 2 mg/dL (RR: 0.4–1.0 mg/dL), AST 200 U/mL (RR: 15–40 U/L), ALT 600 U/mL (RR: 14–50 U/L), ESR 30 mm/hour, CRP 8 mg/dL, ferritin >15,000 ng/mL, triglycerides 500 mg/dL (RR: <500 mg/dL), lactate dehydrogenase 4,000 U/L (RR: 100–200 U/L), sIL-2R 8,000 U/mL (RR: <1,100 U/mL), and absent NK cell activity. CSF studies were normal.
Computed tomography (CT) of the brain was normal. CT of the chest and abdomen revealed bulky lymphadenopathy.
An excisional biopsy of an axillary lymph node showed regressed germinal centers (see Figure 1), hyalinized vascular proliferation (see Figure 2) and mild to moderate plasmacytosis. No hemophagocytosis was noted on the biopsy, staining for HHV-8 was negative, and there were no CD4-/CD8 T cells.
The biopsy was interpreted as angiofollicular lymphoid hyperplasia. Given the pathologist’s comment regarding the Castleman-like features, additional serum laboratory tests, including vascular endothelial growth factor, serum protein electrophoresis, human herpesvirus-8 (HHV-8), and human immunodeficiency virus (HIV) were sent and were unrevealing.
Initially, the working diagnosis was Castleman-like disease secondary to SLE. The patient was managed for macrophage activation syndrome with high-dose corticosteroids and 100 mg of anakinra every six hours, but she was not improving.
Her hospitalization was complicated by oral herpes simplex virus ulcers and Klebsiella bacteremia, which initially limited the choice of additional immunosuppression beyond corticosteroids and anakinra.
Given her lack of response to treatment and the absence of lupus-specific autoantibodies, a multidisciplinary discussion with a hematologist and a pathologist was held. The pivotal question involved whether she had Castleman-like disease secondary to SLE, a key exclusion criterion for idiopathic multicentric Castleman disease (iMCD)—or whether she had primary iMCD.
Diagnosis
Although our patient’s clinical features included fever, hemolytic anemia, altered mental status and diffuse pain, none of these features are specific to SLE. Her renal involvement had included proteinuria; however, she had no significant active urinary sediment, and there was no classic immune complex glomerulonephritis seen on renal biopsy—only podocyte effacement was noted. Nevertheless, her ANA positivity and hypocomplementemia had led providers to favor the diagnosis of SLE during previous evaluations.
Ultimately, we concluded her underlying disease was more consistent with iMCD rather than Castleman-like disease secondary to SLE. She was subsequently given 11 mg/kg of siltuximab, and anakinra was stopped. Following administration of siltuximab, her pancytopenia and inflammatory markers all improved, and we were able to taper corticosteroids.
Our plan was for the patient to receive 11 mg/kg of siltuximab every three weeks; however, she did not follow up after discharge and, ultimately, was lost to care.
Discussion
Castleman disease is a non-neoplastic lymphoproliferative disorder that arises from the clinicopathologic intersection of hematology, oncology, rheumatology and virology.
Unicentric Castleman disease involves a single lymph node station, whereas multicentric Castleman disease (MCD) involves multiple lymph node stations.
HHV-8, occurring most commonly in HIV-infected individuals, is a major driver of MCD, in which viral interleukin (IL) 6 creates a proinflammatory state. In HHV-8-negative MCD, also known as idiopathic MCD (iMCD), the exact etiologies are unknown, but excessive human IL-6 from germinal centers is a pathological driver.
The iMCD diagnostic criteria consist of major criteria, which include multicentric lymphadenopathy and histopathologic lymph node features, such as regressed germinal centers, follicular dendritic cell prominence, vascularity or plasmacytosis. Minor criteria include elevated inflammatory markers, anemia, thrombocytopenia, hypoalbuminemia, renal dysfunction, polyclonal hypergammaglobulinemia, constitutional symptoms, hepatosplenomegaly, anasarca, cherry hemangiomatosis and lymphocytic interstitial pneumonitis. Exclusion criteria include infections, malignant/lymphoproliferative disorders and autoimmune/autoinflammatory diseases, notably SLE.1
Our patient met both major criteria and nine of the 11 minor diagnostic criteria for iMCD. She had a high-titer ANA and hypocomplementemia, but she did not have any lupus-specific autoantibodies or mucocutaneous manifestations of lupus, and her renal, neurologic, hematologic and constitutional manifestations were nonspecific and better explained by iMCD with macrophage activation syndrome.
There are reports of individuals with iMCD that initially mimicked SLE; in one case a patient similarly presented with heterogenous clinical manifestations, nephrotic syndrome and renal biopsy with negative immunofluorescence.2
There are also reports of individuals with SLE and Castleman-like features; in two cases, the Castleman-like features were noted on histological examination of lymph node biopsy, and the patients responded sufficiently to hydroxychloroquine and prednisone.3,4
Conclusion
Although the difference between primary iMCD mimicking SLE vs. SLE with Castleman-like features may be subtle, the correct diagnosis is likely to impact management, and recognition of iMCD in this case led to selection of an IL-6 inhibitor, siltuximab.5 This case underscores the importance of re-evaluating the diagnosis when a patient does not respond to initial treatment. Just as the microscopic examination of histopathology is crucial to the diagnosis, a microscopic examination of clinical details is paramount to the care of the patient. This is the art of medicine.
 Philip Chu, MD, RhMSUS, is a rheumatologist at Duke University Hospital, Durham, N.C., with an interest in musculoskeletal ultrasound.
Philip Chu, MD, RhMSUS, is a rheumatologist at Duke University Hospital, Durham, N.C., with an interest in musculoskeletal ultrasound.
 Mithu Maheswaranathan, MD, is a third-year rheumatology fellow in the Division of Rheumatology at Duke University School of Medicine, Durham, N.C., with interests in health literacy in rheumatic diseases and patient education. [Twitter: @MithuRheum]
Mithu Maheswaranathan, MD, is a third-year rheumatology fellow in the Division of Rheumatology at Duke University School of Medicine, Durham, N.C., with interests in health literacy in rheumatic diseases and patient education. [Twitter: @MithuRheum]
 Jadee L Neff, MD, PhD, is an assistant professor of pathology who subspecializes in hematopathology and molecular genetic pathology at Duke University School of Medicine, Durham, N.C. She is interested in biomarker discovery and assay development for diagnosis and therapeutic management of hematologic malignancy and mimics.
Jadee L Neff, MD, PhD, is an assistant professor of pathology who subspecializes in hematopathology and molecular genetic pathology at Duke University School of Medicine, Durham, N.C. She is interested in biomarker discovery and assay development for diagnosis and therapeutic management of hematologic malignancy and mimics.
 Rebecca E Sadun, MD, PhD, is an adult and pediatric rheumatologist at Duke University School of Medicine, Durham, N.C., with an interest in how cognitive errors impact diagnostic reasoning in rheumatology.
Rebecca E Sadun, MD, PhD, is an adult and pediatric rheumatologist at Duke University School of Medicine, Durham, N.C., with an interest in how cognitive errors impact diagnostic reasoning in rheumatology.
Disclosures
Dr. Neff is a consultant for EUSA Pharma and a participant in the company’s Peer to Peer Speaker’s Bureau regarding the criteria for pathologic diagnosis of Castleman disease.
References
- Fajgenbaum DC, Uldrick TS, Bagg A, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood. 2017 Mar;129(12):1646–1657.
- Wang L, Chen H, Shi J, et al. Castleman disease mimicking systemic lupus erythematosus: A case report. Medicine. 2018 Sep;97(38):e12291.
- Demirkan FG, Doğan S, Kalyoncu Uçar A, et al. Systemic lupus erythematosus complicated with Castleman disease: A case-based review. Rheumatol Int. 2021 Feb;41(2):475–479.
- Xia J-Y, Chen X-Y, Xu F, Yang Y, Wang H-Y, Xue J. A case report of systemic lupus erythematosus combined with Castleman’s disease and literature review. Rheumatol Int. 2012 Jul;32(7):2189–2193.
- Rhee F van, Voorhees P, Dispenzieri A, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood. 2018 Nov;132(20):2115–2124.