Based on her symptoms, her father took her to the emergency department for evaluation. The patient had no recollection of the morning’s events and reported regaining her orientation and awareness on the way to the hospital.
On arrival at the emergency department, she was afebrile and tachycardic (HR 100 beats per minute), with a blood pressure of 125/75 mmHG. She was breathing normally, with oxygen saturation of 99% on room air.
When the patient was admitted to the medical ward, her vision loss had resolved. The physical exam was notable for normal mental status, right upper extremity mild dysmetria, with 4/5 strength in the right upper extremity and right lower extremity, with normal sensation and tone. There was no evidence of meningitis or focal neurologic signs.
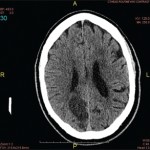
Imaging was obtained and included a computed tomography (CT) of her head, which showed several lesions suspicious for acute or subacute infarction.
Magnetic resonance imaging (MRI) of her brain showed widespread foci of restricted diffusion with corresponding low apparent diffusion coefficient (ADC) signal throughout both cerebral and cerebellar hemispheres, most suggestive of acute infarcts. The distribution was atypical for multiple sclerosis, so they were not thought to be demyelinating plaques, despite her past history. Scattered T2/FLAIR signal hyperintensities noted within the left cerebellum and right cerebral peduncle were suggestive of remote infarcts (see Figures 1 and 2).
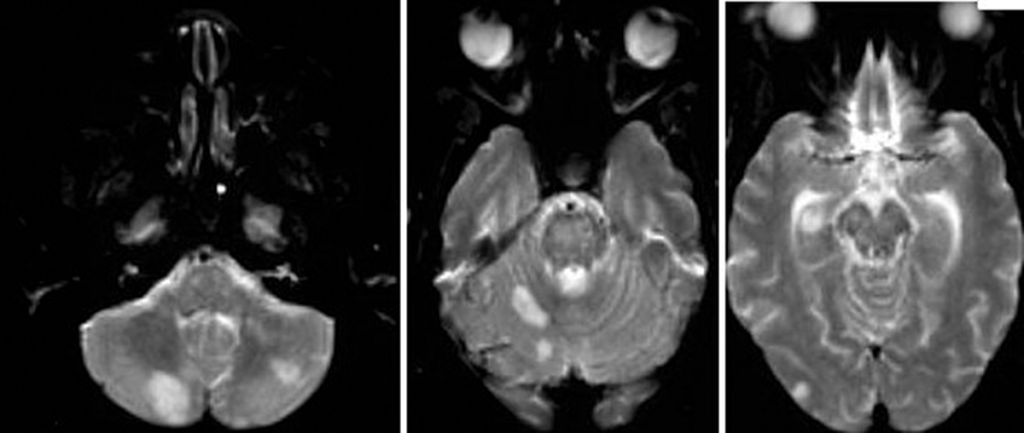
Figure 1A
A magnetic resonance angiogram (MRA) of the head/neck showed no high-grade stenosis, focal occlusion or aneurysm. A 2D echocardiogram showed a normal ejection fraction at 60–65% with diastolic dysfunction and no evidence of masses, thrombi or vegetations.
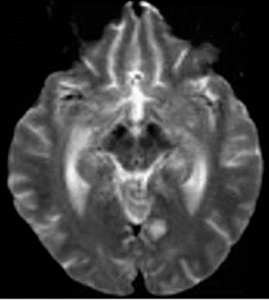
Figure 1B
An ultrasound of the carotid arteries showed 1–39% stenosis of the right and left internal carotid artery, with mild homogeneous plaque.
The patient was started on dual antiplatelet therapy and a statin, and her care was then transferred to the stroke team for continued workup.
The patient’s mental status deteriorated during admission. She became disoriented to questioning and eventually lost the ability to speak, swallow and walk.
Laboratory tests revealed: erythrocyte sedimentation rate of 102 mm/hour (normal: 0–20 mm/hour), C-reactive protein of 1.7 mg/L (normal: <8.2) and hemoglobin of 10.4 mg/dL (normal: 12.0–16.0 mg/dL). The white blood cell count was normal at 8,310/uL (normal: 3,900–12,700/uL), as were her platelets at 250,000/uL (normal: 150,000–350,000/uL). Liver function tests and blood glucose levels were normal. Her creatinine was 3.7 (baseline 2.2), with a glomerular filtration rate of 17 mL/min/1.73 m2 (normal: >60 mL/min/1.73 m2).
She had a Glasgow Coma score (GCS) of 9 (eye opening, 4; verbal response, 1; motor response, 4) and global aphasia with no usable speech or auditory comprehension. Decerebrate posturing was noted during the exam.
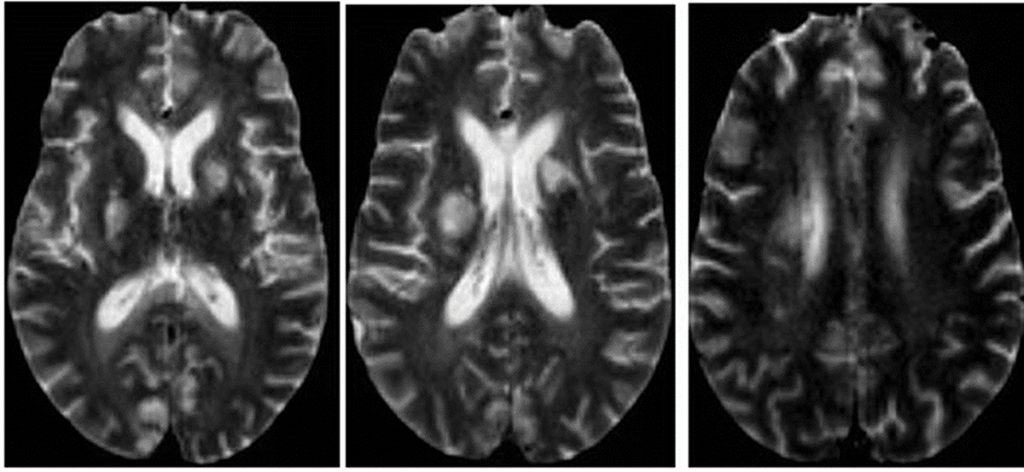
Figure 2: An MRI of the brain without contrast.
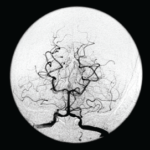
Further tests were performed, including an electroencephalogram (EEG), which was abnormal with generalized slowing indicative of a marked, diffuse, nonfocal and nonspecific encephalopathy. An interventional radiology cerebral angiogram was conducted and showed multifocal areas of distal intracranial artery narrowing, suggestive of vasculitis (see Figures 3 and 4).
A rheumatologist was consulted to evaluate the patient for central nervous system (CNS) vasculitis.
The patient developed acute hypoxic respiratory failure and experienced a febrile episode. She was subsequently intubated and started on broad-spectrum antibiotics (vancomycin and piperacillin/tazobactam). A sputum culture grew Citrobacter werkmanii, which was treated with a seven-day course of piperacillin/tazobactam.
Further lab test results: ANCA was normal (<1:20); MPO-ANCA was 2 units (normal: ≤20 units); PR3-ANCA was <0.2 units (normal: <0.4 (negative) units); ANA was negative (<1:160); anti-NMO antibody was negative; serum cryoglobulins were absent; anti-CCP antibody was 0.5 units/mL (normal: <5.0 units/mL); anti-beta 2 glycoprotein antibody was <9 SGU (normal: ≤20 SGU); DRVVT was negative; anticardiolipin antibodies were <9.40 (normal: 0.00–12.49 MPL); HIV was negative; RPR was non-reactive; a hepatitis panel was negative, as were a TB test and a test for Strongyloides antibodies. Cerebrospinal fluid was colorless, and demonstrated the following: The white blood cell count was 2/mm3 (normal: 0–5/mm3), and 100% of these cells were lymphocytes. The red blood cell count was 60/mm3 (normal: 0/cu mm), and all studies for infectious etiologies were negative, including EBV <2.6 log copies/mL (normal 2.6–7.6 log copies/mL), HSV, CMV PCR <2.30 log IU/mL (normal: <2.30 log IU/mL), Lyme antibody, Aspergillus antibody of <1:8 and Cryptosporidium Ag CSF.
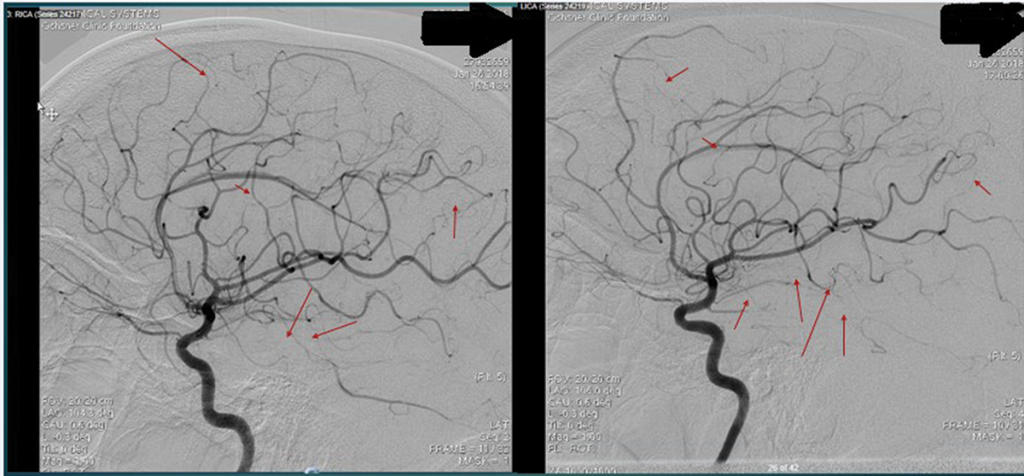
Figure 3 and Figure 4
An interventional radiology angiogram shows multifocal areas of distal intracranial artery narrowing.
Her rheumatoid factor was 22 IU/mL (normal: 0.0–15.0 IU/mL). Her C4 was elevated at 61 mg/dL (normal: 11–44 mg/dL). Her protein/creatinine ratio was 11.07 mg/mmol (normal: 0.00–0.20 mg/mmol).
Several diseases can mimic PACNS, including reversible cerebral vasoconstriction syndrome, premature intracranial atherosclerosis, fibromuscular dysplasia, secondary cerebral vasculitis, malignancy, infections …
Her renal function worsened after the angiogram, and she eventually required dialysis. A nephrologist was consulted and attributed her proteinuria and worsened renal dysfunction to underlying diabetes and contrast-induced nephropathy. A renal ultrasound was unremarkable.
The patient was initially started on 70 mg of prednisone daily. She was switched to pulse-dose steroids (1,000 mg methylprednisolone daily for three days) due to worsening neurologic status. Despite the pulse-dose steroids, the patient’s clinical progression continued to worsen. Repeat imaging showed evolving subacute infarcts.
After the patient was cleared by an infectious disease specialist and after discussion with the neurologist, the decision was made to administer renally dosed cyclophosphamide (415 mg/m2).
About one week after the cyclophosphamide infusion, the patient was able to follow commands and was attempting to state her name. She self-extubated and had an improved GCS of 15.
A brain biopsy showed lymphocytic vasculitis of uncertain etiology and remote cortical microinfarcts, with the affected
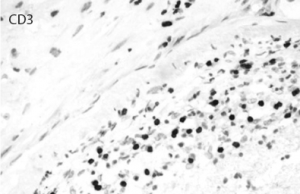
vessels showing a modest number of intramural lymphocytes and foci of chronic inflammatory cells within the adventitia, which were primarily positive for CD3 (see Figures 5–10, right). There was no evidence of granulomatous inflammation, fibrinoid necrosis of the vessel walls or infectious organisms. The patient was tapered to 15 mg methylprednisolone for eight days and eventually discharged to an inpatient rehab facility for follow-up with the rheumatologist, neurologist and nephrologist.
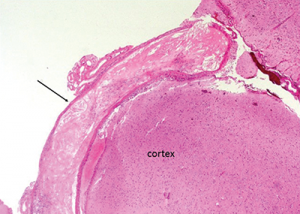
Figure 5: The leptomenigeal artery has significant narrowing due to cholesterol accumulation, and intramural and perivascular inflammatory infiltrates.
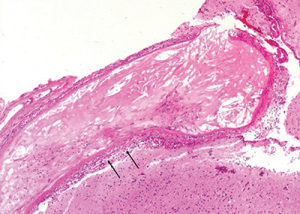
Figure 6: The image at higher magnification is highlighted to show the cholesterol accumulation, and intramural and perivascular inflammatory infiltrates.
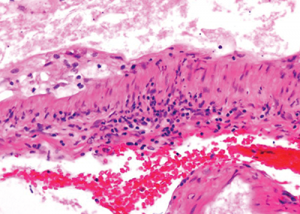
Figure 7: The image at even higher magnification shows the cholesterol accumulation, and intramural and perivascular inflammatory infiltrates.
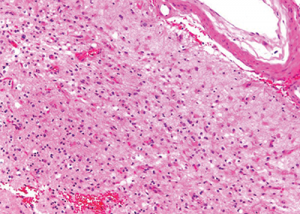
Figure 9: Subacute cortical microinfarct displaying loss of parenchyma and reactive gliosis.
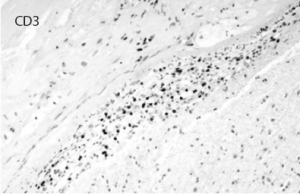
Figure 8: Immunostaining for CD3 highlights the inflammatory T cell population permeating the vascular wall. CD20 was mostly negative.
Courtesy of Maria-Beatriz Lopes, MD, University of Virginia Pathology
Discussion
PACNS is a poorly understood entity, and significant challenges remain for diagnosis and treatment. The pathogenesis is not well understood, and to date no evidence has been found to suggest a genetic predisposition. Various viral infections have been proposed as factors, such as varicella zoster virus, West Nile virus, Mycoplasma gallisepticum and HIV.6-14
In 1988, Calebrese and Mallek proposed diagnostic criteria for PACNS combining clinical, imaging and histology, which includes 1) acquired neurological deficit unexplained after complete evaluation, 2) diagnostic cerebral angiogram with narrowing of vessels suggestive of vasculitis, and 3) no evidence of systemic vasculitis or any other condition that could mimic the angiogram findings.15
These criteria were revised in 2009 by Birnbaum and Hellmann to either a definite or probable diagnosis of PACNS, with the main difference being confirmation of vasculitis on biopsy specimen to prevent patients with reversible vasoconstriction syndromes (RVCS) from being treated with cytotoxic therapy.16
Our patient fulfilled all three criteria for diagnosis of PACNS and had lymphocytic perivascular infiltrates on brain biopsy.
The characteristic histopathologic findings consist of inflammatory infiltration of vessel walls by T lymphocytes and activated macrophages, which undergo granulomatous differentiation with giant cell formation.
Different clinical subsets of PACNS include granulomatous angiitis of the central nervous system (GACNS), a rare subset of PACNS characterized by granulomatous angiitis confined to the brain. Patients present clinically with chronic insidious headaches, along with diffuse and focal neurologic deficits, often bilateral infarcts, as well as high-intensity T2-weighted fluid attenuation inversion recovery (FLAIR) lesions on MRI in the subcortical white matter and deep gray matter.
Atypical CNS vasculitis, another subset of PACNS, comprises multiple manifestations of PACNS that are clinically, radiologically or pathologically distinct from GACNS. The most frequent and heterogeneous subset of PACNS included in this subset are patients with specific presentations, such as lesions, or patients with pathologic findings of lymphocytic infiltration or necrotizing vasculitis with transmural fibrinoid necrosis rather than granulomatous angiitis.17
Benign angiopathy of the central nervous system (BACNS), which is now referred to as reversible cerebral vasoconstriction syndrome, was considered to be the third mimic of PACNS although not a true vasculitis.2
The median age of onset is 50 years, but PACNS may affect patients of all ages. The neurological manifestations are diverse, ranging from hyperacute to chronic and insidious. In a study of 101 patients, Salvarani et al. found patients who presented with subacute manifestations of diffuse CNS dysfunction and acute presentation were highly unusual.18
The most common initial symptoms are headache (63%) and cognitive impairment (50%). Focal symptoms usually appear later in the course of the disease and include hemiparesis (44%), stroke (40%), aphasia (28%), transient ischemic attack (28%), ataxia (19%), seizures (16%), dysarthria (15%) and blurred vision or decreased visual acuity (11%). Infrequent manifestations, occurring in less than 10% of patients, include intracranial hemorrhage; amnesic syndrome; and spinal cord manifestations, such as paraparesis, quadriparesis, Parkinsonism, vertigo, dizziness or cranial nerve palsy; and most patients have multiple manifestations.
No definite laboratory abnormality is diagnostic. The diagnosis of PACNS is challenging because of the nonspecific clinical presentation, the lack of highly specific laboratory and radiologic tests, and the difficulty of obtaining pathologic material. In the majority of published case series of PACNS, abnormal cerebrospinal fluid (CSF) findings have been reported; both CSF pleocytosis and protein elevation are frequent.19 Obtaining appropriate CSF cultures, microbiologic stains, cytology and flow cytometry is crucial to rule out infectious and neoplastic disease.
MRI is the neuroimaging modality of choice for patients with suspected PACNS, and MRI findings are abnormal in 90–100% of patients. MRI may demonstrate ischemic and hemorrhagic lesions of different ages, leukencephalopathies, tumor-like lesions or gadolinium enhancement of the meninges.20 Infarcts may be seen in approximately 50% of cases; when present, infarcts are usually seen bilaterally in multiple-vessel tributaries affecting the cortex, as well as the subcortex. Gadolinium enhancement may occur in as many as one-third of cases; leptomeningeal enhancement may occur in 10–15% of cases, and when present in the nondominant lobe, may represent an ideal site for biopsy.21
Angiography sometimes demonstrates bilateral vessel stenoses or occlusions consistent with an angiitis. In some cases, patients with histologically proved PACNS have an entirely normal angiogram. The angiographic pattern considered diagnostic of vasculitis is often caused by reversible vasoconstriction syndromes associated with drugs, migraine, hypertension, eclampsia or the postpartum period.19,22
Patients must be evaluated for systemic vasculitis, which is characterized by the presence of constitutional symptoms and serological markers indicating systemic inflammation. The inflammatory process in PACNS is limited to the CNS.
Several diseases can mimic PACNS, including reversible cerebral vasoconstriction syndrome, premature intracranial atherosclerosis, fibromuscular dysplasia, secondary cerebral vasculitis, malignancy, infections (e.g., VZV, HIV, hepatitis C, tuberculosis), other multi-system inflammatory disorders, such as sarcoidosis, moyamoya disease, genetic conditions (e.g., CADASIL), posterior reversible encephalopathy syndrome (PRES), chronic hypertension (microvascular cerebral ischemia), demyelinating diseases and others with multifocal cerebral thromboembolism. Other rare syndromes resembling cerebral vasculitis include reversible posterior leukoencephalopathy syndrome, MELAS (mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes), malignant intravascular lymphomatosis, Degos disease, amyloid angiopathy, Fabry’s disease, pseudoxanthoma elasticum, lipohyalinosis and storage diseases.2,20
Brain biopsy is the gold standard for diagnosis to confirm and exclude other etiologies. In a study done by Alrawi et al., out of 61 patients referred for biopsy to test for suspected PACNS, alternative diagnoses were established in 24 cases (39%); 12 patients had infections, including three patients with brain abscesses, and eight patients had malignant neoplasms, including six with primary CNS lymphoma.23 CNS vasculitis is a patchy disease that affects vessels in a skipped and segmental pattern, which limits the sensitivity of the procedure, and as many as 25% of the biopsies were falsely negative.23 To improve the diagnostic yield, targeted biopsies should be performed with inclusion of leptomeninges.17,24
Unfortunately, no randomized studies of PACNS have been done, and thus, all information on treatment is based on retrospective observational data and clinical experience.
Treatment recommendations are derived from the protocols for systemic vasculitides with severe organ involvement in which a combination of steroids with cyclophosphamide is recommended.20 The majority of case series suggest a high degree of good outcomes when patients are treated with glucocorticoids or glucocorticoids and cyclophosphamide.
After remission is achieved, maintenance therapy with azathioprine or mycophenolate mofetil—and, rarely, methotrexate, given its low CNS penetration—is initiated.2 Some reports indicate that combining mycophenolate mofetil and steroids allows disease control with disappearance of the neurological abnormalities, restoration of normal daily activities and dramatic improvement in brain MRI abnormalities.25 Some case reports used rituximab as the initial treatment for CNS vasculitis, and TNF alpha inhibitors are being used for treatment of CNS vasculitis resistant to immunosuppressive treatment.18,26,27
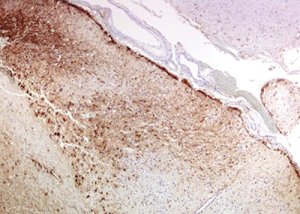
Figure 10: Immunohistochemistry for GFAP (glial fibrillary acidic protein) highlights the reactive gliosis of the subacute infarct.
The disease is considered in remission when the patient is stable and improved clinical and radiologic features are observed. Serial MRIs should be obtained to help in the assessment of disease activity, and clinicians may consider repeat CSF analysis.2
Efforts to limit accelerated atherosclerosis through aggressive control of diabetes, hypertension and dyslipidemia, as well as smoking cessation, is important. Routine vaccinations, osteoporosis prevention and prophylaxis for opportunistic infections should be included in treatment plan.2
The estimated mortality rate of PACNS varies between 10% and 17%. Hajj-Ali et al. found that survival correlated with the initial findings of infarcts and gadolinium-enhanced lesions on MRI.28
In the study analysis of 101 patients, Salvarani et al. reported 20% of patients with PACNS had moderate to severe disability, as assessed by the modified Rankin disability scores and the Barthel index, respectively.5
A literature review shows a Marburg variant of multiple sclerosis that presents with vasculitis and extensive demyelination; however, our patient did not have any areas of demyelination on pathological specimens, which makes this diagnosis less likely.29
A case of stroke occurring after initiation of interferon beta treatment for relapsing-remitting white matter disease has been reported.30 This case is similar to our patient, who had met criteria for multiple sclerosis and developed progression of neurological symptoms after initiation of the interferon-beta treatment. The question remains if her diagnosis of multiple sclerosis was accurate or if her symptoms at that time were related to undiagnosed CNS vasculitis.
Some evidence suggests autoimmune disease, especially vasculitis, worsens with interferon treatment. Thus, another concern is whether the interferon beta treatment was inappropriate and contributed to the severe cerebrovascular insufficiency.30
Conclusion
Diagnosis of PACNS remains a challenge and requires a high index of clinical suspicion, with appropriate workup to exclude other conditions. Further research needs to be done to improve our understanding of the etiology and pathogenesis of this disease, as well as our diagnosis and treatment approaches.
Gbemisola Olayemi, MD, is a graduate of the University of Pretoria, South Africa, and obtained her medical degree at the Medical University of the Americas, Charlestown, Nevis, West Indies. Her internal medicine residency was at Leonard J. Chabert Medical Center, Houma, La., and she is currently engaged in a rheumatology fellowship at Ochsner Clinic Foundation, New Orleans.
Eve Scopelitis, MD, joined the Department of Rheumatology at Ochsner Clinic in 2006. Prior to that she was professor of medicine and director of the Rheumatology Clinics at the Louisiana State University Health Sciences Center, New Orleans. Her medical degree was obtained at SUNY Upstate Medical Center in Syracuse, N.Y. She completed an internal medicine residency at Nassau County Medical Center, East Meadow, N.Y., and a fellowship in Rheumatology at the LSU Health Sciences Center, New Orleans.
Jerald M. Zakem, MD, joined the Department of Rheumatology at Ochsner Clinic in 2000. He is currently an Instructor for the University of Queensland at Ochsner Clinical School. Prior to that, he was a clinical assistant professor of medicine at the University of South Florida in Tampa. His medical degree was obtained at University of Louisville, Ky. He completed an internal medicine residency and a rheumatology fellowship at the University of South Florida.
References
- Carolei A, Sacco S. Central nervous system vasculitis. Neurol Sci. 2003 May;24(suppl 1):S8–S10.
- Firestein G, Budd R, Gabriel SE, et al. (2012). Kelley’s Textbook of Rheumatology, 9th ed. Philadelphia: Saunders, an imprint of Elsevier.
- Harbitz F. Unknown forms of arteritis with special reference to their relation to syphilitic arteritis and periarteritis nodosa. Am J Med Sci. 1922;163:250–272.
- Cravioto H, Feigin I. Noninfectious granulomatous angiitis with a predilection for the nervous system. Neurology. 1959 Sep;9:599–609.
- Salvarani C, Brown RD Jr., Calamia KT, et al. Primary central nervous system vasculitis: Analysis of 101 patients. Ann Neurol. 2007 Nov;62(5):442–451.
- Sigal LH. The neurologic presentation of vasculitic and rheumatologic syndromes. A review. Medicine (Baltimore). 1987 May;66(3):157–180.
- van der Ven AJ, van Oostenbrugge RJ, Kubat B, Tervaert JW. Cerebral vasculitis after initiation antiretroviral therapy. AIDS. 2002 Nov 22;16(17):2362–2364.
- Kossorotoff M, Touzé E, Godon-Hardy S, et al. Cerebral vasculopathy with aneurysm formation in HIV-infected young adults. Neurology. 2006 Apr 11;66(7):1121–1122.
- Thomas L, Davidson M, McCluskey RT. Studies of PPLO infection. I. The production of cerebral polyarteritis by Mycoplasma gallisepticum in turkeys; the neurotoxic property of the Mycoplasma. J Exp Med. 1966 May 1;123(5):897–912.
- Outteryck O, Sénéchal O, Berteloot D, et al. [Cerebral vasculitis secondary to Varicella-Zoster virus infection]. Rev Neurol (Paris). 2005 Sep;161(8–9):836–839.
- Alexander JJ, Lasky AS, Graf WD. Stroke associated with central nervous system vasculitis after West Nile virus infection. J Child Neurol. 2006 Jul;21(7):623–625.
- Brannagan TH 3rd. Retroviral-associated vasculitis of the nervous system. Neurol Clin. 1997 Nov;15(4):927–944.
- Bhat G, Mathur DS, Saxena GN, et al. Granulomatous angiitis of the central nervous system associated with herpes zoster. J Assoc Physicians India. 2002 Jul;50:977–978.
- Nogueras C, Sala M, Sasal M, et al. Recurrent stroke as a manifestation of primary angiitis of the central nervous system in a patient infected with human immunodeficiency virus. Arch Neurol. 2002 Mar;59(3):468–473.
- Calabrese LH, Mallek JA. Primary angiitis of the central nervous system. Report of 8 new cases, review of the literature, and proposal for diagnostic criteria. Medicine (Baltimore). 1988 Jan;67(1):20–39.
- Birnbaum J, Hellmann DB. Primary angiitis of the central nervous system. Arch Neurol. 2009 Jun;66(6):704–709.
- Miller DV, Salvarani C, Hunder GG, et al. Biopsy findings in primary angiitis of the central nervous system. Am J Surg Pathol. 2009 Jan;33(1):35–43.
- Salvarani C, Brown RD Jr., Huston J 3rd, et al. Treatment of primary CNS vasculitis with rituximab: Case report. Neurology. 2014 Apr 8;82(14):1287–1288.
- Schmidley JW. (2000) Central Nervous System Angiitis. Boston: Butterworth-Heinemann.
- Berlit P. Diagnosis and treatment of cerebral vasculitis. Ther Adv Neurol Disord. 2010 Jan;3(1):29–42.
- Pomper MG, Miller TJ, Stone JH, et al. CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography. AJNR Am J Neuroradiol. 1999 Jan;20(1):75–85.
- Calabrese LH, Dodick DW, Schwedt TJ, Singhal AB. Narrative review: Reversible cerebral vasoconstriction syndromes. Ann Intern Med. 2007 Jan 2;146(1):34–44.
- Alrawi A, Trobe JD, Blaivas M, Musch DC. Brain biopsy in primary angiitis of the central nervous system. Neurology. 1999 Sep 11;53(4):858–860.
- Calabrese LH, Furlan AJ, Gragg LA, Ropos TJ. Primary angiitis of the central nervous system: Diagnostic criteria and clinical approach. Cleve Clin J Med. 1992 May–Jun;59(3):293–306.
- Salvarani C, Brown RD Jr., Christianson TJ, et al. Mycophenolate mofetil in primary central nervous system vasculitis. Semin Arthritis Rheum. 2015 Aug;45(1):55–59.
- De Boysson H, Arquizan C, Guillevin L, Pagnoux C. Rituximab for primary angiitis of the central nervous system: Report of 2 patients from the French COVAC cohort and review of the literature. J Rheumatol. 2013 Dec;40(12):2102–2103.
- Salvarani C, Brown RD Jr., Calamia KT, et al. Efficacy of tumor necrosis factor alpha blockade in primary central nervous system vasculitis resistant to immunosuppressive treatment. Arthritis Rheum. 2008 Feb 15;59(2):291–296.
- Hajj-Ali R, Villa-Forte AI, Abou-Chebel A, et al. Long term outcome of patients with primary angiitis of the central nervous system (PACNS). Arthritis Rheum. 2000 Sep;43(suppl 9):S162.
- Elenein RG, Sharer LR, Cook SD, et al. A second case of Marburg’s variant of multiple sclerosis with vasculitis and extensive demyelination. Mult Scler. 2011 Dec;17(12):1531–1538.
- Bosche B, Dohmen C, Impekoven P, et al. Stroke after initiation of interferon‐beta treatment for relapsing–remitting disseminated white matter disease. Acta Neuro Scand. 2006 Jun;113(6);433–436.