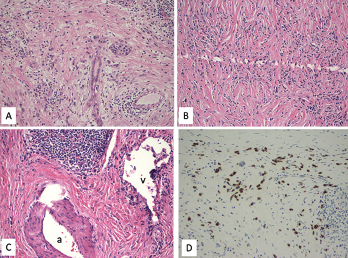
Figure 1. A. Lymphoplasmacytic infiltrate with pancreatic exocrine component atrophy. There is a cluster of remaining islet cells in the pancreatic duct. H&E magnification x200; B. Dense fibrosis with storiform pattern. H&E magnification x200; C. Lymphoplasmacytic infiltrate of a vein (phlebitis) without obliteration of the lumen. H&E magnification x200; v—vein, a—artery; D. Immunostain for IgG4 highlights increased IgG4-positive plasma cells, up to 80 cells/HPF. Immunostain, magnification x200.
Sarcoidosis and IgG4-related disease (IgG4-RD) are both immune-mediated, often multi-organ, diseases of uncertain etiology capable of presenting with diverse clinical manifestations. Many clinical features are common to both conditions, including hypergammaglobulinemia, the ability to form inflammatory masses and involvement of the lymph nodes, lacrimal glands, salivary glands, meninges and lungs. Although imaging modalities, such as positron emission tomography/computed tomography (PET/CT), and serologic evaluation may suggest one disease over the other, these diagnoses remain dependent on tissue sampling and histopathologic examination. Histopathologic evaluation is also essential to rule out other mimicking conditions, such as lymphoma.
Here, we present the case of a patient previously diagnosed with IgG4-RD, after presenting with a pancreatic mass, who was subsequently diagnosed with sarcoidosis based on a biopsy of a mediastinal lymph node.
With the current presentation, a comprehensive review of systems did not uncover evidence of fever, chills, sweats and weight loss. Her family history was negative for psoriasis, inflammatory bowel disease and inflammatory arthritis. She denied any smoking history, but admitted to social alcohol use.
On physical exam, she was in no distress and had normal vital signs. Her salivary glands were enlarged, but not tender. Multiple non-blanching urticarial lesions were seen over both arms, without involvement of her legs. The musculoskeletal exam was normal, with no evidence of synovitis. A lung exam was clear throughout, and a cardiovascular exam revealed a regular heart rate and rhythm without murmurs, and normal pulses.
The initial laboratory evaluation revealed thrombocytosis with platelets of 517,000/uL (reference range [RR]: 140,000–400,000/uL), a white blood cell count of 6,000/uL (RR: 3,800–10,800/uL), hemoglobin of 14 g/dL (RR: 11.7–15.5 g/dL) and hematocrit of 42% (RR: 35.0–45.0%). She had an elevated total cholesterol level of 266 mg/dL (normal <200 mg/dL) and increased fasting glucose of 162 mg/dL (RR: 65–99 mg/dL). The C-reactive protein (CRP) was elevated at 14.3 mg/L, but the erythrocyte sedimentation rate (ESR) was normal at 8 mm/hr (normal: <30 mm/hr). The complete metabolic profile, including chemistries and liver function, was within normal ranges. Amylase, lipase, calcium and vitamin D metabolites were within normal ranges.
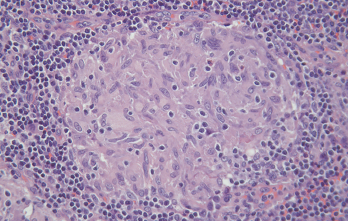
Figure 2. A non-caseating granuloma from mediastinal lymph node biopsy confirming sarcoidosis. H&E magnification x200.
To further characterize the salivary gland enlargement and to evaluate for the possibility of more extensive disease, the patient underwent a PET/CT. These studies showed numerous areas of lymphadenopathy in the cervical chain, axilla, mediastinum and retroperitoneum, measuring up to a maximum of 6.53 standardized uptake values (SUV) and measuring up to 1.2 cm in diameter.
An excisional biopsy of a mediastinal lymph node revealed non-caseating granulomatous changes with negative stains for fungi and acid-fast bacilli (see Figure 2, opposite). This pathology was most consistent with sarcoidosis. Given her medical history of probable IgG4-RD, the specimen was reviewed by a second pathologist internally and also sent for external consultation.
The consensus was for a diagnosis of sarcoidosis, citing the characteristic cellular architecture for sarcoidosis with the presence of non-caseating granulomas and lack of significant numbers of IgG4-positive plasma cells on immunostaining.
The patient was treated for sarcoidosis with glucocorticoids and methotrexate as a steroid-sparing agent. She initially experienced partial relief in the intensity of the urticarial lesions, along with modest improvement in the CRP elevation and reduction in the size of the salivary
gland lymphadenopathy.
With persistent constitutional symptoms of fatigue, ongoing rash and elevated CRP, the patient was started on adalimumab 40 mg subcutaneously every two weeks to help halt the systemic inflammation. However, she did not see significant improvement in her skin changes with the addition of adalimumab, nor was there any decrease of her blood glucose and CRP levels, and the decision to stop the medication was made.
Discussion
Sarcoidosis is an inflammatory disease that can affect any organ system to varying degrees. Although it has been seen in a wide array of populations, it is most commonly reported in Black women, particularly affecting adults under the age of 40 years, and peaking between 20 and 29 years of age.
Patients may report symptoms related to the specific organs involved, and diagnosis must be made on the basis of the encompassing clinical presentation, histological and radiological findings, as well as exclusion of other similarly presenting diseases.
The lungs are most commonly involved, and patients may present with dry cough, dyspnea and chest pain. Imaging classically shows bilateral hilar lymphadenopathy, and pathology shows non-caseating granulomas that can occur in lymph nodes, perivascular sheaths and connective tissue spaces. Because sarcoidosis can mimic several other diseases, it is imperative to rule out other diagnoses, including malignancy, granulomatosis with polyangiitis and infectious causes.1,2
IgG4-RD is a fibroinflammatory disease that, similar to sarcoidosis, can affect nearly any part of the body. Whereas sarcoidosis most often involves the lungs and lymph nodes, IgG4-RD tends to affect the lacrimal glands, salivary glands and pancreas most frequently.
Several markers have been studied in the diagnosis of IgG4-RD, including serum IgG4 levels, plasmablasts and complement levels, which alone are not specific for the diagnosis.3 Histopathology typically demonstrates lesions with elevated IgG4-expressing plasma cells, along with a dense lymphoplasmacytic infiltrate, fibrosis in a storiform pattern and obliterative phlebitis.4
New classification criteria for IgG4-RD were published by the ACR and EULAR in 2019.5 These criteria are composed of three main parts that focus on typical organ involvement, relevant exclusion criteria and sufficient points for classification based on a scoring system that takes clinical, radiologic and histopathologic aspects into account.
Although sarcoidosis and IgG4-RD have some similar characteristics, they remain unique and complex diseases capable of presenting alone or concurrently. Two case reports have been published describing patients presenting with both diseases.
In the first case, an 80-year-old woman presenting with chronic diarrhea and weight loss was found to have pancreatic enlargement on imaging, with evidence of autoimmune pancreatitis on biopsy. Paired with a serum IgG4 concentration of 680 mg/dL (normal: <80 mg/dL), the presentation was strongly supportive of IgG4-RD. Concurrently, a lymph node biopsy showed non-caseating granulomas, consistent with sarcoidosis and inconsistent with IgG4-RD.6
In the second case, a 36-year-old African American man with previously diagnosed sarcoidosis presented with bilateral periorbital swelling. CT imaging showed enlarged lacrimal glands that were then biopsied and histologically supported a diagnosis of IgG4-RD, with findings of dense fibrous tissue, lymphoplasmacytic inflammation and an increased number of IgG4-expressing plasma cells.7
Treatment for sarcoidosis centers on the use of glucocorticoids.1 Conflicting data support the use of methotrexate, hydroxychloroquine or tumor necrosis factor-α (TNF-α) inhibitors for sarcoidosis.8 For IgG4-RD, treatment with glucocorticoids is also first-line therapy. Rituxumab is a preferred treatment for refractory cases, while limited available data suggest the efficacy of immunosuppressive agents, such as azathioprine and mycophenolate.9
In our case, the patient was originally started on prednisone with only partial efficacy. She was started on methotrexate, followed by adalimumab, with the goal of decreasing her systemic inflammatory response from sarcoidosis.
The treatment of IgG4-RD with TNF-α inhibitors has only been reported in case reports, with the hypothesis that a reduction in TNF-α mediated fibrosis may reduce disease activity.
The cases: A 61-year-old woman with IgG4-RD orbital involvement was treated with infliximab, and a 9-year-old girl with IgG4-RD lacrimal gland involvement was treated with adalimumab.10,11 Both of these patients had been resistant to previous therapies. We recommend using the most rigorous classification criteria available to aid in the choice of treatment.
For our patient, the choice of adalimumab treatment after the initial failure of glucocorticoids and methotrexate held the potential to treat both sarcoidosis and IgG4-RD with a medication that was safe and easily discontinued due to its dosing frequency, unlike the irreversible effect of rituximab in depleting B cells. However, after three months of treatment, we did not observe significant changes in her acute phase reactants and the decision to stop treatment was made. The patient deferred further treatment with rituximab.
Because the clinical features of sarcoidosis and IgG4-RD can overlap, a histological diagnosis is critical. This approach also helps rule out other important mimics of the two diseases, such as malignancy or infection.12 There have been limited comparisons of the two diseases at the cellular and cytokine level.
In patients with pulmonary sarcoidosis and IgG4-RD respiratory disease, the bronchoalveloar lavage (BAL) fluid of patients was compared. The cytokine profile of sarcoidosis patients was predominately that of Th1 cytokines with higher levels of interleukin (IL) 2, IL-6 and TNF-α. The IgG4-RD patients had higher levels of Th2 cytokines IL-5 and IL-13.13 These studies demonstrate how far away we stand from the ability to differentiate between IgG4-RD and sarcoidosis with a molecular signature, despite the similar clinical features they may share.
Our case highlights the importance of considering alternative and mimicking diagnoses in patients with a history of IgG4-RD who present with lympha denopathy.
 Veronica Matto, DO, began a fellowship in rheumatology at Larkin Community Hospital, Miami, in July 2021.
Veronica Matto, DO, began a fellowship in rheumatology at Larkin Community Hospital, Miami, in July 2021.
 Rajshri Shah, MD, is an associate pathologist with Comprehensive Pathology Associates PA and is affiliated with Baptist Hospital of Miami.
Rajshri Shah, MD, is an associate pathologist with Comprehensive Pathology Associates PA and is affiliated with Baptist Hospital of Miami.
 Jie Ouyang, MD, PhD, is a pathologist for AdventHealth Orlando, Florida, and member of the United States and Canadian Academy of Pathology.
Jie Ouyang, MD, PhD, is a pathologist for AdventHealth Orlando, Florida, and member of the United States and Canadian Academy of Pathology.
 Cory Perugino, DO, is a rheumatologist and physician-scientist at Massachusetts General Hospital, Boston, where he focuses on translational and basic immunology research pertinent to IgG4-related disease.
Cory Perugino, DO, is a rheumatologist and physician-scientist at Massachusetts General Hospital, Boston, where he focuses on translational and basic immunology research pertinent to IgG4-related disease.
 Joseph LaConti, MD, PhD, is a rheumatologist at the Center for Arthritis and Rheumatic Diseases, Miami, and member of the American Arthritis and Rheumatology Associates.
Joseph LaConti, MD, PhD, is a rheumatologist at the Center for Arthritis and Rheumatic Diseases, Miami, and member of the American Arthritis and Rheumatology Associates.
Disclosures
Dr. LaConti is a member of the AbbVie and GlaxoSmithKline speakers bureaus.
References
- Thomas KW, Hunninghake GW. Sarcoidosis. JAMA. 2003 Jun 25;289(24):3300–3303.
- Costabel U, Hunninghake GW. ATS/ERS/WASOG statement on sarcoidosis. Sarcoidosis Statement Committee. American Thoracic Society. European Respiratory Society. World Association for Sarcoidosis and Other Granulomatous Disorders. Eur Respir J. 1999 Oct;14(4):735–737.
- Khosroshahi A, Wallace ZS, Crowe JL, et al. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheumatol. 2015 Jul;
67(7):1688–1699. - Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Modern Pathology. 2012 Sep;25(9):1181–1192.
- Wallace Z, Naden R, Chari S, et al. The 2019 American College of Rheumatology/European League Against Rheumatism classification criteria for IgG4-related disease. Arthritis Rheumatol. 2020 Jan;72(1):7–19.
- Michel L, Clairand R, Néel A, et al. Association of IgG4-related disease and sarcoidosis. Thorax. 2011 Oct;66(10):920–921.
- Fogt F, Wellmann A, Lee V. Immunoglobulin G4-related disease (IgG4-RD) with associated sarcoidosis. J Ocular Biol. 2013;1(1):3.
- Iannuzzi, MC, Rybicki, BA, Teirstein, AS. Sarcoidosis. N Engl J Med. 2007 Nov 22;357(21):2153–2165.
- Khosroshahi A, Stone JH. Treatment approaches to IgG4-related systemic disease. Curr Opin Rheumatol. 2011 Jan;23(1):67–71.
- Karim F, Paridaens D, Westenberg LEH, et al. Infliximab for IgG4-related orbital disease. Ophthalmic Plast Reconstr Surg. 2017 May/Jun;33(3S Suppl 1):S162–S165.
- Jalaj S, Dunbar K, Campbell A, Kazim M. Treatment of pediatric IgG4-related orbital disease with TNF-α anhibitor. Ophthalmic Plast Reconstr Surg. 2017 Jan/Feb;
34(1):e10–e12. - Wessendorf TE, Bonella F, Costabel U. Diagnosis of sarcoidosis. Clin Rev Allergy Immunol. 2015 Aug;49(1):54–62.
- Yamamoto H, Yasuo M, Ichiyama T, et al. Cytokine profiles in the BAL fluid of IgG4-related respiratory disease compared with sarcoidosis. ERJ Open Res. 2015 Oct 23;1(2):00009-2015.