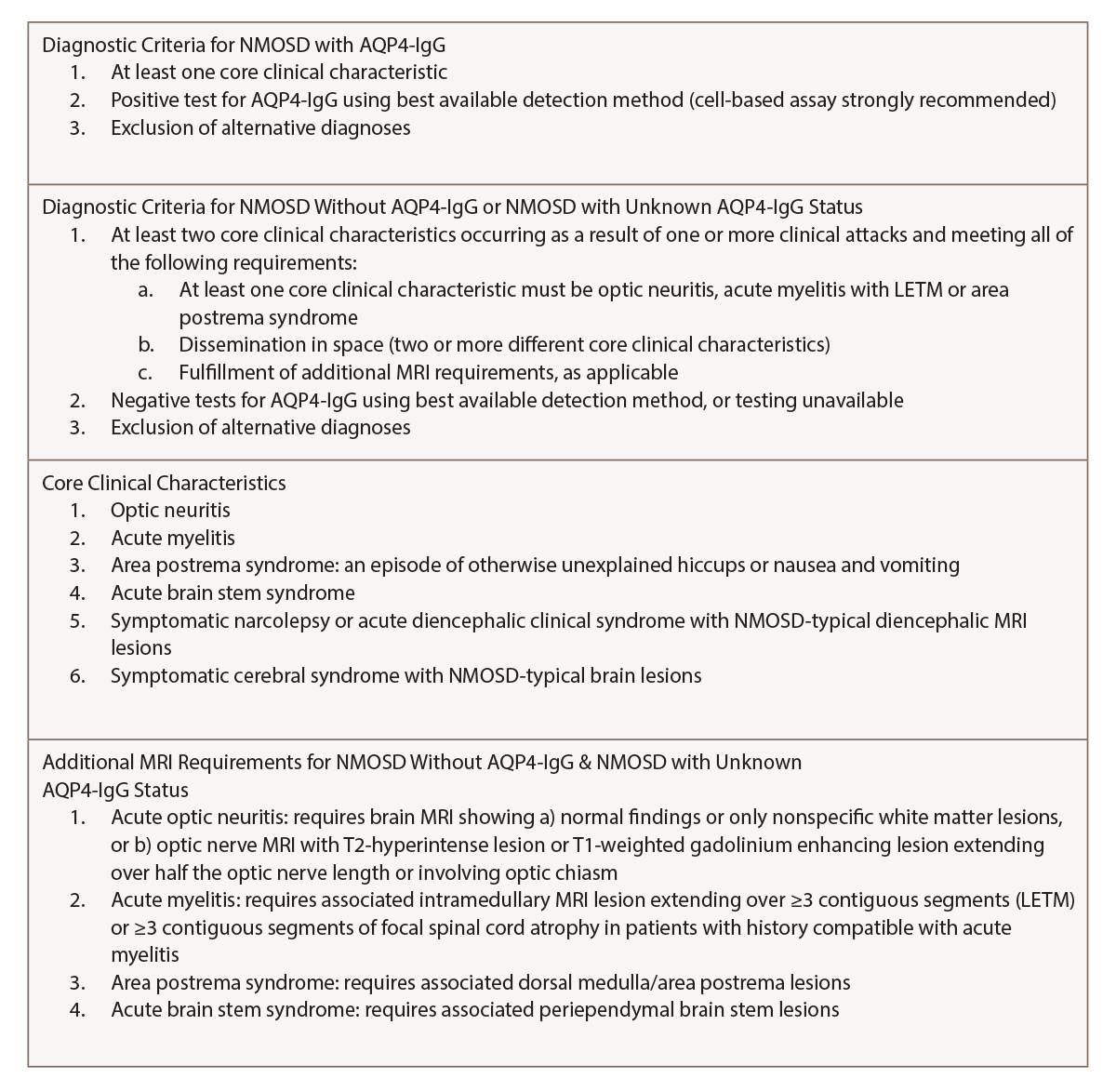
(click for larger image) Table 1: NMOSD Diagnostic Criteria for Adults by the IPND1
Discussion
Diagnostic Criteria
NMOSD is a rare autoimmune demyelinating disease of the CNS. It is characterized by episodes of unilateral or bilateral optic neuritis and longitudinally extensive (LETM; three or more vertebral segments) transverse myelitis, as well as brain stem and hypothalamic lesions.1 It is also known as Devic’s disease, because Eugene Devic was the first to provide a systematic description of it in 1894.2
NMOSD is distinct from multiple sclerosis (MS) and is associated with an anti-aquaporin-4 IgG antibody that targets AQP4 channels, the primary CNS water channel that is expressed on the astrocytic foot processes. Studies have suggested this antibody may play a role in the disease pathogenesis. It appears to be involved in the destruction of AQP4 channels on the astrocyte foot processes by complement-dependent cytotoxicity, cytokine release and blood-brain barrier disruption. This leads to oligodendrocyte death and the observed neurological manifestations of NMOSD.3 The presence of anti-AQP4 antibodies is associated with either a relapsing or refractory disease course. However, it does not correlate with disease activity, so it can’t be used as a marker of response to therapy.4
AQP4-IgG serology was incorporated into NMO diagnostic criteria in 2006. In 2007, the term neuromyelitis optica spectrum disorder was introduced to include isolated unilateral or bilateral recurrent optic neuritis, as well as recurrent transverse myelitis and myelitis associated with autoimmune conditions, such as Sjögren’s syndrome and systemic lupus erythematosus (SLE).5
The diagnostic criteria were revised by the IPND for NMOSD in 2015 to include both NMOSD with and without AQP4-IgG. The IPND-consensus diagnostic criteria for NMOSD appear in Table 1 (p. 21). In the presence of anti-AQP4 IgG, one of the six core clinical characteristics is needed for the diagnosis. However, patients who lack AQP4-IgG must have two or more core clinical characteristics with other MRI findings.1
NMOSD in Sjögren’s Syndrome
A variety of neurologic manifestations may occur as a result of Sjögren’s syndrome. The actual pathogenesis of CNS involvement in primary Sjögren’s syndrome remains unclear, but autoimmune-mediated demyelination is a proposed pathogenic mechanism. Studies have found that the AQP4-IgG antibody is seen exclusively in patients with NMOSD, suggesting this is a separate entity and not a direct CNS manifestation of Sjögren’s syndrome.6
Diagnosis
Zekeridou et al. indicated an elevated anti-AQP4-IgG antibody has a sensitivity of approximately 80% and specificity greater than 95% in the diagnosis of NMOSD.7 Myelin oligodendrocyte glycoprotein (MOG) antibody may be present in NMOSD patients with negative anti-AQP4-IgG antibody.8 CSF fluid analysis is critical to exclude infections. CSF in 50% of NMOSD patients contains elevated white blood cell counts during acute flares. CSF protein may be elevated. IgG oligoclonal bands in CSF, which are the hallmark of MS, are typically absent in NMOSD.9
Neuroimaging
Spinal MRI reveals increased signal on sagittal T2-weighted STIR sequences extending over three or more complete vertebral segments and gadolinium enhancement on T1-weighted sequences. Typical NMOSD lesions are large, continuous and located centrally in the corticospinal tract. Lesions may also appear in the hemispheric cerebral white matter, the periependymal region in the diencephalon, the dorsal brain stem and the white matter adjacent to lateral ventricles. In contrast, MS lesions are small, ovoid, present in the cortical and juxtacortical region, and perpendicularly oriented to the surface of the lateral ventricle.
Orbital MRI shows increased T2 signal and gadolinium enhancement of the optic nerve or optic chiasm, suggestive of optic neuritis. Optical coherence tomography (OCT) may show severe damage to the retinal nerve fiber layer and the ganglion cell layer following attacks of optic neuritis in NMOSD.10