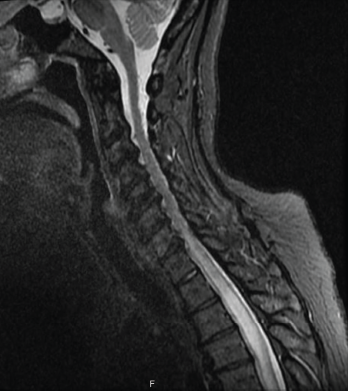
Figure 1: MRI of the spine
Sjögren’s syndrome is a chronic multi-system autoimmune disease characterized by inflammation and subsequent destruction of exocrine glands. Sjögren’s syndrome can present with glandular or extra-glandular manifestations. Neuromyelitis optica spectrum disorder (NMOSD) is a rare central nervous system (CNS) autoimmune disease that can present as the initial manifestation in less than 5% of patients with Sjögren’s.
It is important to recognize relationships between autoimmune diseases, especially Sjögren’s syndrome and NMOSD. This rare association requires a high degree of suspicion in a patient with Sjögren’s syndrome with a suggestive clinical presentation and imaging findings.
On physical examination, she had bilateral lower extremity flaccid paralysis, absent bilateral patellar and Achilles tendon reflexes, and loss of sensation for pain, temperature, light touch, vibration and joint position sense below the T4 level. All other system reviews were unremarkable.
Magnetic resonance imaging (MRI) of the spine indicated intra- and extramedullary spinal cord enhancement from T3–T11, and edema from C5–T11 (see Figure 1). An MRI of the brain revealed a 2 cm T1 hypointense, T2/FLAIR hyperintense periventricular lesion in the left frontoparietal centrum semiovale with mild perivascular enhancement. An orbital MRI suggested an increased signal within the right optic nerve without focal enhancement, consistent with optic neuritis (see Figure 2).
Her cerebrospinal fluid (CSF) analysis showed a white blood cell count of 1,300/uL (normal: 0–5/uL) with 88% neutrophils and elevated CSF protein of >300 mg/dL (normal: 15–45 mg/dL). Other CSF studies—including acid-fast bacilli, bacterial and fungal culture, venereal disease research lab, herpes simplex virus, cryptococcus, West Nile virus, Epstein-Barr virus, toxoplasma, blastomyces, histoplasma, cytomegalovirus and varicella tests—were negative. CSF oligoclonal bands, serum and CSF lymphoma panels, and a positron emission tomography scan were also normal. Serum aquaporin-4 (AQP4) immunoglobulin G antibodies (IgG) were elevated at 362.1 U/mL (normal: 0–3 U/mL).
Based on the 2015 revised criteria by the International Panel for NMO Diagnosis (IPND, see Table 1), a diagnosis of NMOSD was made.
The patient was initially treated with intravenous dexamethasone and tapered to 40 mg of oral prednisone daily. She received IV antibiotics due to an initial concern for infection, which was discontinued when her CSF culture proved negative. Additionally, she underwent five cycles of plasmapheresis. Her weakness did not improve during hospitalization, so she was transitioned to acute rehab to follow up in the rheumatology clinic for initiation of subcutaneous tocilizumab.
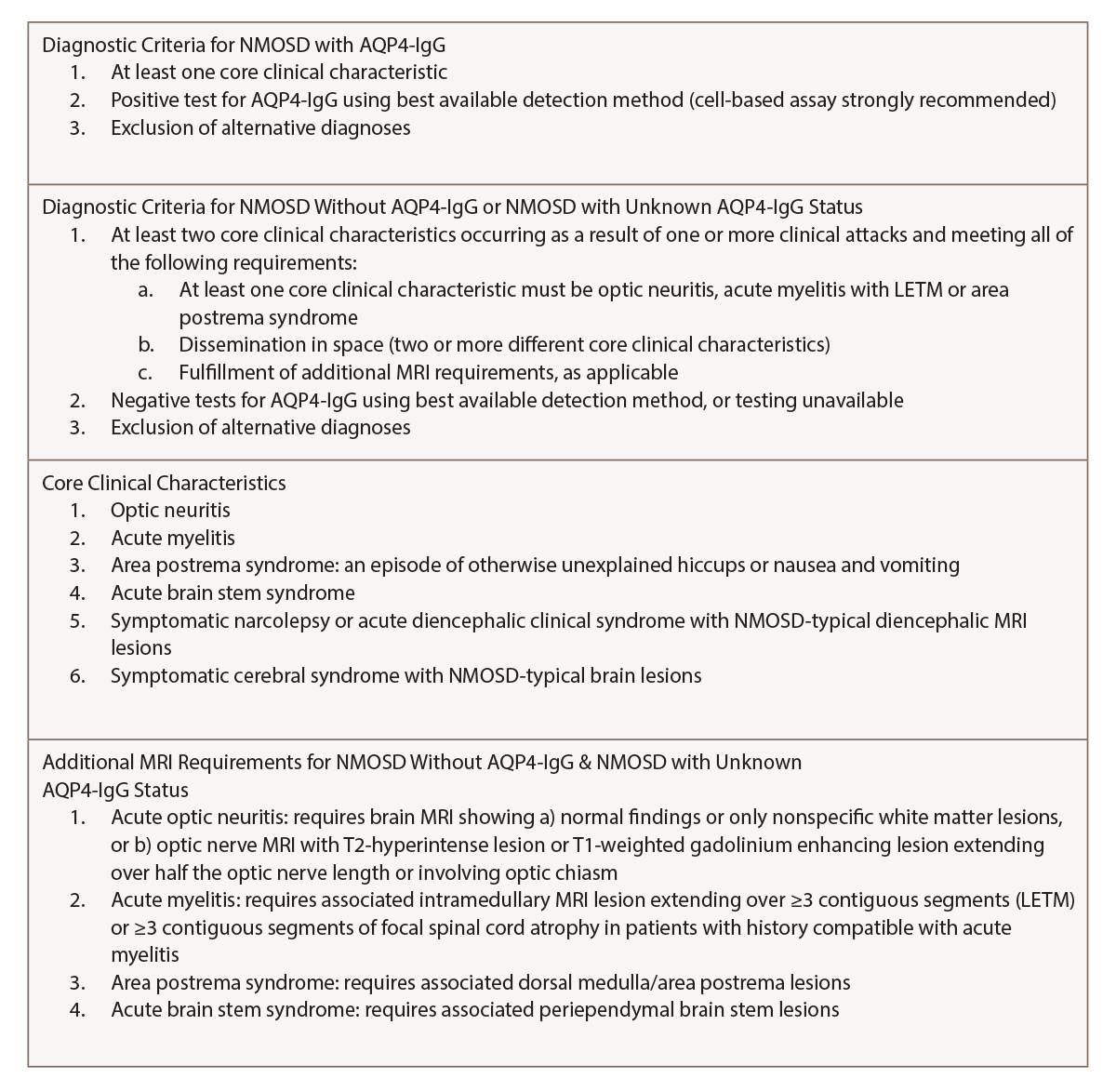
(click for larger image) Table 1: NMOSD Diagnostic Criteria for Adults by the IPND1
Discussion
Diagnostic Criteria
NMOSD is a rare autoimmune demyelinating disease of the CNS. It is characterized by episodes of unilateral or bilateral optic neuritis and longitudinally extensive (LETM; three or more vertebral segments) transverse myelitis, as well as brain stem and hypothalamic lesions.1 It is also known as Devic’s disease, because Eugene Devic was the first to provide a systematic description of it in 1894.2
NMOSD is distinct from multiple sclerosis (MS) and is associated with an anti-aquaporin-4 IgG antibody that targets AQP4 channels, the primary CNS water channel that is expressed on the astrocytic foot processes. Studies have suggested this antibody may play a role in the disease pathogenesis. It appears to be involved in the destruction of AQP4 channels on the astrocyte foot processes by complement-dependent cytotoxicity, cytokine release and blood-brain barrier disruption. This leads to oligodendrocyte death and the observed neurological manifestations of NMOSD.3 The presence of anti-AQP4 antibodies is associated with either a relapsing or refractory disease course. However, it does not correlate with disease activity, so it can’t be used as a marker of response to therapy.4
AQP4-IgG serology was incorporated into NMO diagnostic criteria in 2006. In 2007, the term neuromyelitis optica spectrum disorder was introduced to include isolated unilateral or bilateral recurrent optic neuritis, as well as recurrent transverse myelitis and myelitis associated with autoimmune conditions, such as Sjögren’s syndrome and systemic lupus erythematosus (SLE).5
The diagnostic criteria were revised by the IPND for NMOSD in 2015 to include both NMOSD with and without AQP4-IgG. The IPND-consensus diagnostic criteria for NMOSD appear in Table 1 (p. 21). In the presence of anti-AQP4 IgG, one of the six core clinical characteristics is needed for the diagnosis. However, patients who lack AQP4-IgG must have two or more core clinical characteristics with other MRI findings.1
NMOSD in Sjögren’s Syndrome
A variety of neurologic manifestations may occur as a result of Sjögren’s syndrome. The actual pathogenesis of CNS involvement in primary Sjögren’s syndrome remains unclear, but autoimmune-mediated demyelination is a proposed pathogenic mechanism. Studies have found that the AQP4-IgG antibody is seen exclusively in patients with NMOSD, suggesting this is a separate entity and not a direct CNS manifestation of Sjögren’s syndrome.6
Diagnosis
Zekeridou et al. indicated an elevated anti-AQP4-IgG antibody has a sensitivity of approximately 80% and specificity greater than 95% in the diagnosis of NMOSD.7 Myelin oligodendrocyte glycoprotein (MOG) antibody may be present in NMOSD patients with negative anti-AQP4-IgG antibody.8 CSF fluid analysis is critical to exclude infections. CSF in 50% of NMOSD patients contains elevated white blood cell counts during acute flares. CSF protein may be elevated. IgG oligoclonal bands in CSF, which are the hallmark of MS, are typically absent in NMOSD.9
Neuroimaging
Spinal MRI reveals increased signal on sagittal T2-weighted STIR sequences extending over three or more complete vertebral segments and gadolinium enhancement on T1-weighted sequences. Typical NMOSD lesions are large, continuous and located centrally in the corticospinal tract. Lesions may also appear in the hemispheric cerebral white matter, the periependymal region in the diencephalon, the dorsal brain stem and the white matter adjacent to lateral ventricles. In contrast, MS lesions are small, ovoid, present in the cortical and juxtacortical region, and perpendicularly oriented to the surface of the lateral ventricle.
Orbital MRI shows increased T2 signal and gadolinium enhancement of the optic nerve or optic chiasm, suggestive of optic neuritis. Optical coherence tomography (OCT) may show severe damage to the retinal nerve fiber layer and the ganglion cell layer following attacks of optic neuritis in NMOSD.10
The actual pathogenesis of CNS involvement in primary Sjögren’s syndrome remains unclear, but autoimmune-mediated demyelination is a proposed pathogenic mechanism.
The differential diagnosis for the neurological manifestations found in Sjögren’s syndrome is extensive and includes infections (e.g., HIV, syphilis, fungal, bacterial and viral), malignancy, sarcoidosis and other demyelinating syndromes, such as MS and idiopathic transverse myelitis. Serum calcium, angiotensin converting enzyme levels and chest X-ray should be obtained, because sarcoidosis can present with optic
neuropathy and myelopathy. CNS lymphoma should be excluded by performing a CSF and serum lymphoma panel. However, it is important to recognize that intravascular CNS lymphoma may prove difficult to exclude.
Treating Acute Episodes
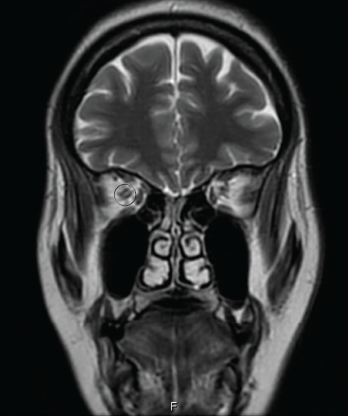
Figure 2: AN ORBITAL MRI
The goals of acute treatment are to suppress the acute inflammatory attack, minimize CNS damage and maintain long-term neurological function. Pulse-dose corticosteroids (e.g., 1,000 mg of IV methylprednisolone) daily for five days, followed by a two- to six-month taper, serve as initial therapy. Plasmapheresis has shown to be effective in patients who are refractory or show minimal response to steroids, making it a good alternative in a patient with coexisting infections.11
Long-Term Management
Azathioprine (AZA) was the first preventive therapy studied in 1998. Subsequent studies confirmed the initial observation of the utility of AZA (3 mg/kg/day) and also indicated the combination of prednisone plus AZA yielded improved outcomes.12
Three studies published from 2009–2014 suggest mycophenolate was more effective in achieving remission in 60–75% of subjects with fewer side effects than azathioprine.13
From 2005–2015, multiple prospective and retrospective rituximab studies indicated sustained remission in up to 83% of NMOSD patients. Further, rituximab was superior to either mycophenolate or azathioprine. Rituximab dosing is either 375 mg/m2 weekly for four doses, or 1,000 mg every two weeks for a total of two doses, followed by scheduled infusions every six months.13
Tocilizumab, an interleukin (IL) 6 inhibitor, has been studied in two pilot studies, because IL-6 is found to be elevated in the blood and spinal fluid of relapsing and active NMOSD patients.14 In the first study, performed in Japan in seven NMOSD patients, tocilizumab (8 mg/kg/month) decreased the relapse rate from 2.9 to 0.4 when added to background immunosuppressive therapy, such as azathioprine or prednisone. IL-6 inhibition also improved neuropathic pain and general fatigue.15 These findings were confirmed in a study performed in Germany in eight NMOSD patients.16
Eculizumab, a C5 inhibitor that blocks the terminal activation of complement and the membrane attack complex, and CD19 monoclonal antibody (MEDI-551), are under investigation to evaluate their ability to prevent NMOSD relapses.12
New treatments have been proposed that target a specific component of disease pathogenesis, including aquaporumab, a recombinant human monoclonal antibody that is composed of an Fc portion binding to AQP4 and thus reduces AQP4-IgG binding, and sivelestat, a neutrophil elastase inhibitor involved in neutrophil migration and phagocytosis.13
NMOSD is a rare autoimmune demyelinating disease of the CNS. It is characterized by episodes of unilateral or bilateral optic neuritis & longitudinally extensive (three or more vertebral segments) transverse myelitis, as well as brain stem & hypothalamic lesions.
Conclusion
NMOSD is a rare, demyelinating, inflammatory disorder that can coexist in patients with Sjögren’s syndrome. It is a separate clinical diagnosis and not a direct neurological manifestation of Sjögren’s syndrome. The presence of an anti-AQP4-IgG antibody helps in the diagnosis. Immunosuppressive therapy, such as mycophenolate, azathioprine and rituximab, prevents long-term neurological sequelae, but plasmapheresis is preferred if there is any concern for coexisting infections. IL-6 inhibitors can be used in patients with active or relapsing NMOSD.
Swosti Roka Magar, MD, is a rheumatology fellow at the University of Arkansas for Medical Sciences, Little Rock.
Gita Verma, MD, is a rheumatology fellow at the University of Arkansas for Medical Sciences, Little Rock.
Aaroop Haridas, MD, is an assistant professor in the Division of Rheumatology at the University of Arkansas for Medical Sciences, Little Rock.
References
- Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015 Jul 14;85(2):177–189.
- Jarius S, Wildemann B. The history of neuromyelitis optica. J Neuroinflammation. 2013 Jan 15;10:8.
- Ratelade J, Verkman AS. Neuromyelitis optica: Aquaporin-4 based pathogenesis mechanisms and new therapies. Int J Biochem Cell Biol. 2012 Sep;44(9):1519–1530.
- Weinshenker BG, Wingerchuk DM, Vukusic S, et al. Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol. 2006 Mar;59(3):566–569.
- Pandit L. Neuromyelitis optica spectrum disorders: An update. Ann Indian Acad Neurol. 2015 Sep;18(Suppl 1):S11–S15.
- Birnbaum J, Atri NM, Baer AN, et al. Relationship between neuromyelitis optica spectrum disorder and Sjögren’s syndrome: Central nervous system extraglandular disease or unrelated, co-occurring autoimmunity? Arthritis Care Res (Hoboken). 2017 Jul;69(7):1069–1075.
- Zekeridou A, Lennon VA. Aquaporin-4 autoimmunity. Neurol Neuroimmunol Neuroinflamm. 2015 May 21;2(4):e110.
- Kitley J, Waters P, Woodhall M, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: A comparative study. JAMA Neurol. 2014 Mar;71(3):276–283.
- Jarius S, Paul F, Franciotta D, et al. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: Results from 211 lumbar punctures. J Neurol Sci. 2011 Jul 15;306(1–2):82–90.
- Kim HJ, Paul F, Lana-Peixoto MA, et al. MRI characteristics of neuromyelitis optica spectrum disorder: An international update. Neurology. 2015 Mar 17;84(11): 1165–1173.
- Kimbrough DJ, Fujihara K, Jacob A, et al. Treatment of neuromyelitis optica: Review and recommendations. Mult Scler Relat Disord. 2012 Oct;1(4):180–187.
- Kessler RA, Mealy MA, Levy M. Treatment of neuromyelitis optica spectrum disorder: Acute, preventive and symptomatic. Curr Treat Options Neurol. 2016 Jan;18(1):2.
- Sherman E, Han MH. Acute and chronic management of neuromyelitis optica spectrum disorder. Curr Treat Options Neurol. 2015 Nov;17(11):48.
- Içöz S, Tüzün E, Kürtüncü M, et al. Enhanced IL-6 production in aquaporin-4 antibody positive neuromyelitis optica patients. Int J Neurosci. 2010 Jan;120(1):71–75.
- Araki M, Matsuoka T, Miyamoto K, et al. Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: A pilot study. Neurology. 2014 Apr 15;82(15):1302–1306.
- Ringelstein M, Ayzenberg I, Harmel J, et al. Long-term therapy with interleukin 6 receptor blockade in highly active neuromyelitis optica spectrum disorder. JAMA Neurol. 2015 Jul;72(7):756–763.