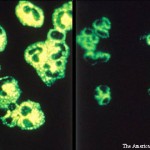
Case
Click to enlarge.
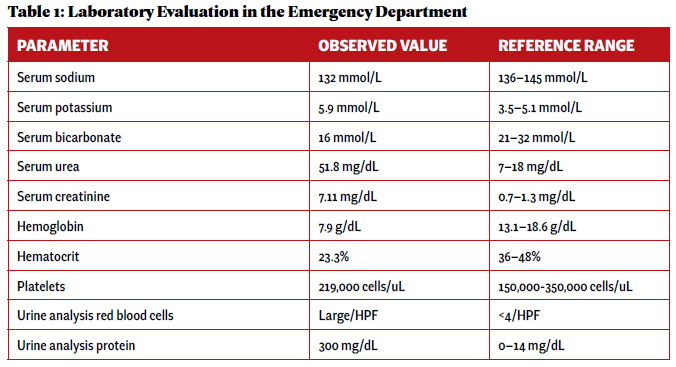
An otherwise healthy 23-year-old woman presented to her primary care provider with gross hematuria initially attributed to her menstrual cycles. Over the next five months, she developed edema, hemoptysis, headaches, nausea and vomiting, and experienced episodes of epistaxis prompting evaluation at an emergency department. Pertinent initial laboratory results on hospital admission are presented in Table 1.
Laboratory evaluation demonstrated acute renal failure, 2+ proteinuria and gross hematuria. In addition, evaluation in the emergency department was significant for multi-focal pulmonary opacities seen on a computed tomography (CT) scan of the chest (see Figure 1).
Her clinical presentation was ultimately consistent with a pulmonary-renal syndrome, with features of diffuse alveolar hemorrhage and renal failure, and she was transferred to our hospital for inpatient consultations with a nephrologist and a rheumatologist.
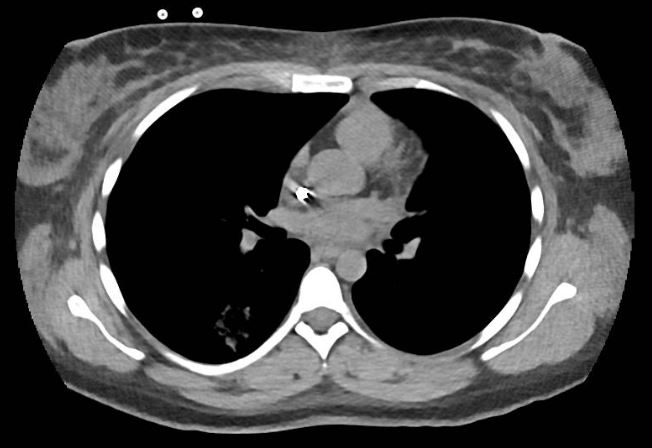
FIGURE 1: Computed tomography of the patient’s chest revealed multi-focal opacities. (Click to enlarge.)
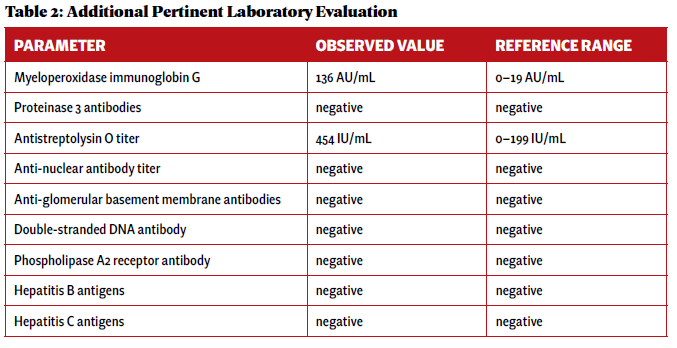
An additional laboratory evaluation was performed at a different hospital to assess the etiology of her pulmonaryrenal syndrome (see Table 2).
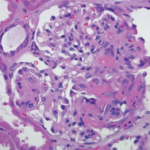
A renal biopsy was performed due to suspected vasculitis. Biopsy results demonstrated a rapidly progressive glomerulonephritis secondary to ANCA-associated vasculitis, with active crescentic and necrotizing glomerulonephritis, 50% sclerosis and some mesangial immunoglobin A (IgA ) deposition. With the presence of multiple crescents in the context of antimyeloperoxidase (anti-MPO) positivity and minimal immunofluorescence deposition, ANCA-associated crescentic glomerulonephritis with superimposed mild IgA nephropathy was favored as a diagnosis.
The patient was ultimately diagnosed with ANCA-associated vasculitis in the setting of MPO positivity and pauci-immune glomerulonephritis, as demonstrated on renal biopsy.
Click to enlarge.
The patient was initiated on pulse-dose corticosteroids, plasmapheresis and supportive dialysis, and 1,000 mg of rituximab separated by 14 days. Seven cycles of plasmapheresis were initially anticipated, but the therapy was stopped after two cycles due to a notable response to the corticosteroid treatment. The patient was discharged home with a corticosteroid taper and continued on supportive dialysis in the outpatient setting.