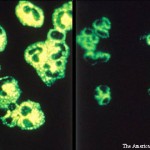
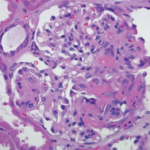
Two weeks after initial hospital discharge, the patient returned to an emergency department complaining of worsening abdominal and bilateral flank pain. Additional imaging was ultimately negative for new thrombosis or infarction, and her symptoms were attributed to volume overload in the setting of dialysis. Laboratory testing incidentally revealed new-onset thrombocytopenia and low-grade schistocytes (see Table 3).
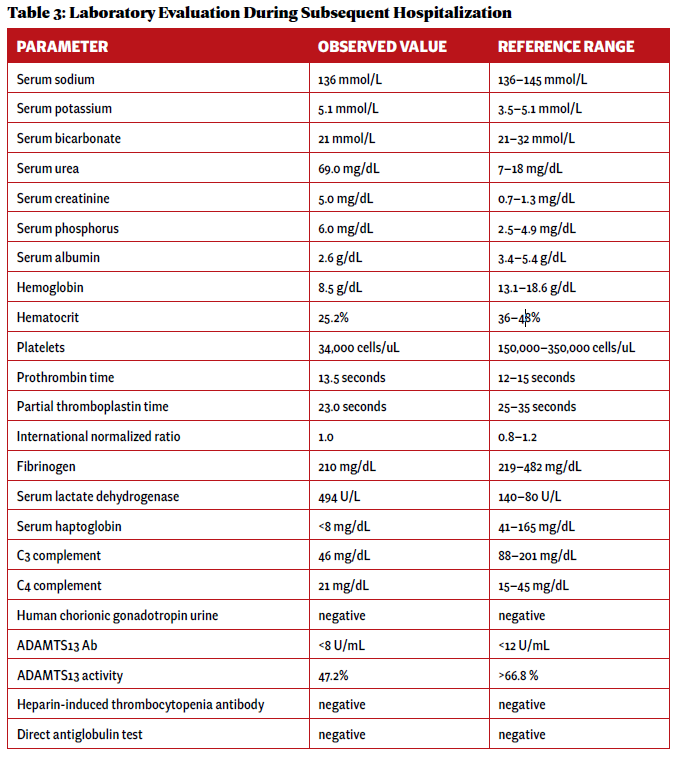
Click to enlarge.
These laboratory findings and her clinical presentation were ultimately consistent with microangiopathy hemolytic anemia and thrombocytopenia, collectively a thrombotic microangiopathy syndrome. In the setting of low complement levels with no additional features of an overlap rheumatic syndrome, a diagnosis of c-TMA was made.