What’s Your Diagnosis?
- Majocchi’s granuloma/tinea infection
- Granuloma annulare
- Palisaded neutrophilic granulomatous dermatitis
- Familial sarcoidosis/Blau syndrome
Correct answer: D
Familial Sarcoidosis/Blau Syndrome
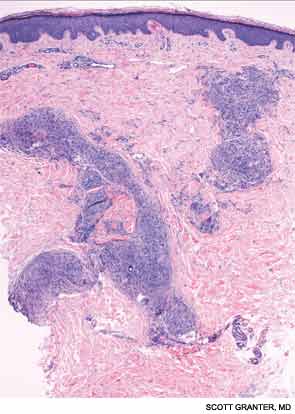
A skin biopsy of a representative lesion was performed, and histopathologic examination revealed nodular aggregates of histiocytes forming non-necrotizing, “naked” granulomas (see Figure 2). Special stains and PCR studies for infectious organisms including mycobacteria were negative. Genetic testing revealed that our patient and multiple family members had a base substitution in the CARD15/NOD2 gene, consistent with a diagnosis of Blau syndrome. Topical steroids were prescribed for her lesions, which resolved with treatment.
Blau syndrome was first described in 1985 by Edward Blau, a pediatrician. It’s a rare, autosomal dominant autoinflammatory disorder with a typical onset within the first two decades of life. Clinically, patients most commonly present with a triad of granulomatous dermatitis, symmetric arthritis and recurrent uveitis. Patients may also have other organ involvement, including hepatic, renal, cardiovascular or neurologic complications.1,2 Mutations that cause Blau syndrome have been mapped to CARD15/NOD2, a caspase recruitment domain gene.
The CARD15/NOD2 gene contains three domains. It is most commonly known for mutations in the third domain, associated with Crohn’s disease, which involves granulomatous inflammation of the bowel. Mutations in the second domain are associated with both Blau syndrome and early onset sarcoidosis. CARD15/NOD2 activates NF-kB in the setting of bacterial cell wall components.3 In Blau syndrome, genetic mutations in the second domain are usually gain-of-function mutations, leading to increased NF-kB activation and possibly increased activity of other inflammatory cytokines.4
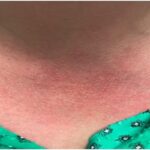
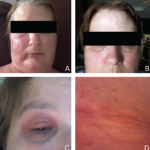
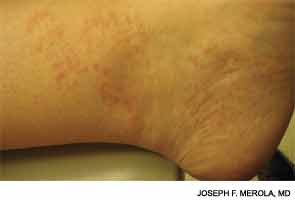
Blau syndrome often presents earliest and most commonly in the skin. Patients present with multiple erythematous and waxy dermal papules and plaques with a lack of scale. Skin biopsy is helpful in diagnosis because it often demonstrates granulomatous inflammation.5
Discussions of Other Choices
Majocchi’s granuloma occurs when a dermatophyte infection involves the deeper structures of the skin, such as the hair follicles and the dermis. These lesions occur commonly in areas with hair follicles, such as the legs. Patients can also demonstrate other evidence of dermatophyte infection, such as onychomycosis or tinea pedis. Histopathologic examination of Majocchi’s granuloma would reveal fungal forms on the biopsy.
Granuloma annulare (GA) commonly presents as an erythematous, annular plaque with no scale. However, the subcutaneous form of GA can present as solitary or multiple deeper dermal nodules. Histopathologic examination of these lesions would demonstrate histiocytes around a focus of necrobiosis and increased mucin deposition, the intervening dermis typically appears normal. Multinucleated giant cells are frequently seen along with mild perivascular and interstitial lymphocytic infiltrate with scattered neutrophils.
Palisaded neutrophilic granulomatous dermatitis (PNGD) presents as umbilicated papules distributed symmetrically on the extremities, particularly extensor extremities, as well as on the hands and feet. PNGD is commonly associated with seropositive rheumatoid arthritis and other autoimmune diseases, including systemic lupus erythematosus. On histopathology, PNGD demonstrates a robust neutrophilic and granulomatous histiocytic infiltrate with collagen degeneration and possibly fibrin deposition. These lesions may be thought of as “rheumatoid papules,” a smaller, more superficial variant of the rheumatoid nodule.
Joseph F. Merola, MD, MMSc, is an instructor at Harvard Medical School, Brigham and Women’s Hospital, in Boston. He is board-certified in rheumatology, dermatology and internal medicine. He serves as the director of clinical trials in dermatology, co-director of the Center for Skin and Related Musculoskeletal Diseases and assistant program director for the combined medicine-dermatology residency training program.
Johanna Sheu, MS, is a medical student at Harvard Medical School. She is currently completing a research fellowship in pediatric dermatology at Boston Children’s Hospital.
References
- Rose CD, Martin TM, Wouters CH. Blau syndrome revisited. Curr Opin Rheumatol. 2011;23:411–418.
- Wang X, Kuivaniemi H, Bonavita G, et al. CARD15 mutations in familial granulomatosis syndromes: A study of the original Blau syndrome kindred and other families with large-vessel arteritis and cranial neuropathy. Arthritis Rheum. 2002;46:3041–3045.
- Inohara N, Ogura Y, Fontalba A, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278:5509–5512.
- Sfriso P, Caso F, Tognon S, et al. Blau syndrome, clinical and genetic aspects. Autoimmunity Reviews. 2012;12:44–51.
- Janssen CE, Rose CD, De Hertogh G, et al. Morphologic and immunohistochemical characterization of granulomas in the nucleotide oligomerization domain 2-related disorders Blau syndrome and Crohn disease. J Allergy Clin Immunol. 2012;129:1076–1084.