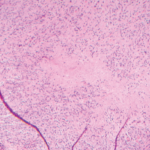
Several groups have now demonstrated that the formation of memory B cells is severely impaired in the majority of CVID patients and leads to a concomitant depletion of long-lived plasma cells and a drop of serum Ig levels. The altered distribution of B cell subsets observed in CVID patients is now applied in disease classification.5-7
Diagnosis
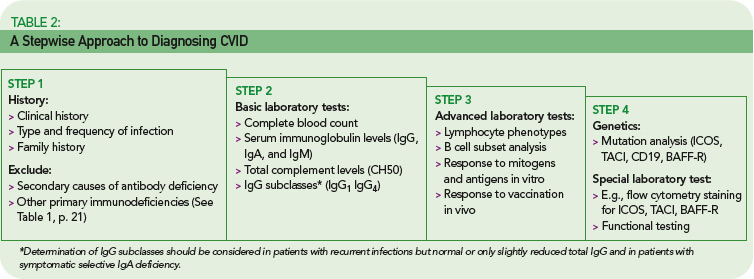
The rare incidence and high clinical variability of CVID can present a diagnostic challenge. Because there is no specific test for diagnosing CVID, diagnosis is made by excluding similar conditions. First, the clinically more frequent secondary causes for hypogammaglobulinemia must be ruled out (See Table 1, below). Then other primary immunodeficiency disorders have to be excluded, and suspected CVID should be validated, as outlined in Table 2 (p. 22).
The first step is the assessment of the clinical and family history, including a detailed description of the type, duration, and frequency of infections. In addition to recurrence of infections, unusual severity and development of complications (e.g., empyema, bronchiectasis) can indicate an underlying immune defect. Identifying underlying pathogens is very helpful in evaluating any immunodeficient patient, with infections by encapsulated bacteria being the most common type in CVID. Whenever possible, use direct detection methods for pathogens (e.g., microbial culture, antigen-ELISA, PCR) to evaluate the cause of infection, because serology is futile in patients with an antibody deficiency.