SG was then transferred to our hospital for further evaluation. On arrival, he was afebrile and vital signs were normal. On neurologic examination, he was fully oriented, but was unable to relay his history due to word-finding difficulties, and speech was fragmented and circumferential in nature. He was able to recall zero out of three objects at five minutes. His cranial nerves were intact, but he had decreased hearing in the right ear. His reflexes were equal and intact. Mild dysmetria was observed with his left upper and lower extremities. His gait was unstable and he could not walk in tandem. His behavior was disinhibited and he expressed paranoid and delusional thoughts.
Additional information about his history was obtained. He worked for a pharmaceutical company and as a personal trainer. He denied smoking, illicit drugs, heavy alcohol consumption, or recent travel. His family history was notable for a father with multiple transient ischemic attacks and a maternal uncle with multiple sclerosis (MS). He had no family history of rheumatologic diseases.
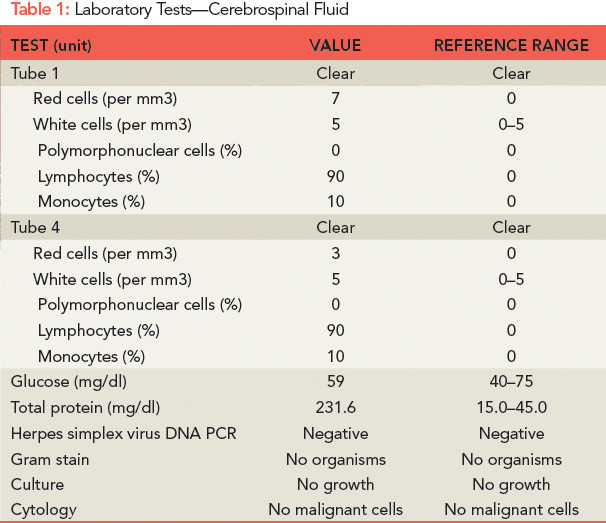
Further laboratory tests were sent (see Table 2). Electroencephalography (EEG) revealed generalized bursts of slowing, consistent with a mild to moderate encephalopathy of toxic, metabolic, or anoxic etiology with no evidence of seizure activity. A repeat MRI of his brain was performed (see Figure 1), which showed innumerable small foci of restricted diffusion scattered over the cerebrum, cerebellum, and brainstem, increased from the prior MRI, with some cortical lesions, but primarily involving the white matter, notably the corpus callosum. The foci were hyperintense on FLAIR imaging and there was persistent leptomeningeal enhancement. There was no evidence of acute infarction or hemorrhage.
Diagnosis
The differential diagnosis at this point was broad. Infectious etiologies were first investigated. Viral etiologies, particularly herpes simplex virus type 1 (HSV-1) encephalitis, characterized by fever, headache, focal neurologic findings, and impaired consciousness, were considered.1 However, SG did not have fevers or a headache, and CSF polymerase chain reaction (PCR), which has very high sensitivity and specificity for detection of HSV-1, was negative.2 Further tests for other infections that can manifest with central nervous system (CNS) involvement, including human immunodeficiency virus (HIV), human T-lymphotropic virus 1 and 2, syphilis, Lyme disease, hepatitis B and C, and tuberculosis, were negative. Toxic ingestions and illicit drugs were also investigated and both his toxicology screen and his serum alcohol level were negative.
Primary neurologic diseases were also considered. Given his initial viral prodrome, the increase in CSF protein, and his MRI findings of multifocal white-matter lesions, post-viral acute disseminated encephalomyelitis (ADEM) was a concern. The possibility of MS was discussed, however his brain MRI findings did not appear to be consistent with this diagnosis, and an MRI of his cervical and thoracic spine showed no evidence of demyelination. Two well-defined gamma-restriction bands were seen in both his CSF and his serum, but this is a nonspecific finding, not suggestive of MS. Creutzfeldt-Jakob disease was considered, given his memory loss and psychiatric symptoms; however, he had no evidence of myoclonus, and did not exhibit rapid mental deterioration.3 Malignancy, especially lymphoma, was plausible given the leptomeningeal enhancement seen on his MRI; CSF cytology, however, was negative for malignant cells.
A rheumatologic disease, most notably primary angiitis of the CNS (PACNS), was another possibility. PACNS has a male predominance, and a mean age of 42 years.4 Clinical manifestations range widely, and include decreased cognition, headache, seizures, and strokes. Normal inflammatory markers, negative antineutrophil cytoplasmic antibodies (ANCA), and a normal MRA do not exclude this diagnosis. SG had no clear history, clinical, or physical manifestations suggestive of systemic autoimmune diseases such as systemic lupus erythematosus, Behçet’s disease, vasculitis, sarcoidosis, or polyarteritis nodosa, which can cause CNS disease. In terms of his laboratory tests, his antinuclear antibody (ANA) was negative on initial check and then low-titer positive (1:80) on repeat. Lupus anticoagulant, beta-2 glycoprotein antibodies, and anticardiolipin IgG were negative. Anticardiolipin IgM was in the indeterminate range, and ANCA was negative. Erythrocyte sedimentation rate was normal and C-reactive protein was very slightly elevated. Standard cerebral angiography was suggested but not performed.
Finally, Susac’s syndrome, or retinocochleocerebral vasculopathy, was considered, given his multifocal MRI findings with involvement of the central fibers of the corpus callosum and leptomeningeal enhancement, in addition to encephalopathy and unilateral hearing loss.5 The finding of branch retinal artery occlusions is central to this diagnosis.
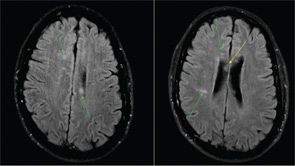
The ophthalmology team was asked to evaluate SG, and a fluorescein angiography of the retina was performed (See Figure 2). A superotemporal branch retinal artery occlusion was seen with focal areas of nonperfusion in the right retina. There were associated areas of perivascular and retinal whitening with cotton wool spots. The occlusion was outside of the macula, which explains the preservation of vision in our patient. Initial imaging of the left retina also revealed an inflamed retinal arteriole; however, the examination could not be completed because of patient noncompliance.
Based on this constellation of clinical and angiographic findings, the diagnosis of Susac’s syndrome was established.
Discussion
Susac’s syndrome, or retinocochleocerebral vasculopathy, was first described in 1979 as a triad of multiple-branch retinal artery occlusions, brain microangiopathy causing encephalopathy, and sensorineural hearing loss.5,6 Since the initial description of this rare syndrome in two young women by John O. Susac, approximately 100 cases have been reported in the medical literature.7 Cases demonstrate a female predominance (3:1) with an age range from 16 to 58 years.6 Susac’s syndrome is described as a “microangiopathy” or a “vasculopathy,” distinct from primary angiitis of the CNS, as only precapillary arterioles are involved.6 Pathogenesis remains unknown, but it is thought to be an immunologically mediated vasculitis of small blood vessels. One case described a microangiopathic syndrome in a patient with features suggestive of systemic lupus erythematosus, although antinuclear antibodies were negative.8 No other links to systemic rheumatologic diseases or to hypercoagulable states have been described. In prior case reports, the encephalopathy, which may be acute or subacute, has manifested with memory loss, dysarthria, ataxic gait, and pronounced psychiatric features, similar to this case.9 Often, patients have headaches, which were not described here. CSF analysis tends to be abnormal with a mild pleocytosis and significantly increased protein level (>100), as was the case with this patient.10
In a clinical pathologic conference presented in the New England Journal of Medicine (NEJM) in 2011, a brain biopsy in a patient with Susac’s syndrome was performed. Findings demonstrated swollen endothelial cells, a small amount of mononuclear-cell perivascular inflammation, and intraluminal hyaline thrombi, suggestive of a microangiopathy.11 There was no evidence of granulomatous angiitis, demyelination, malignancy, or infection. In other cases, brain biopsy has revealed both acute and subacute microinfarcts, and changes in the microvasculature, similar to the NEJM case.11-14
MRI findings in Susac’s syndrome include small, multifocal T2 hyperintensities, most notably in the cerebral white matter and the corpus callosum, and approximately 30% have leptomeningeal enhancement.15 Lesions are hyperintense on FLAIR images. All of the reported cases involve the corpus callosum, usually the central fibers, but spare the periphery, which is distinct from the MRI findings in patients with MS or ADEM.6 This feature was also seen in our patient. Given the small size of the vessels involved, MRA and standard cerebral angiography are normal in patients with Susac’s syndrome. EEG findings are nonspecific.
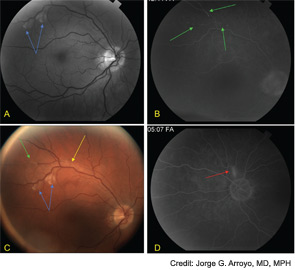
Fluorescein angiography of the retina often shows hyperfluorescence of the wall of the retinal arterioles, and complete occlusion of distal branches.16 The occlusions may be associated with Gass plaques, atheromatous deposits caused by leakage of lipids into the arterial wall at the site of damage.16 Cotton wool patches and boxcar segmentation may also be seen, as was seen in our patient. Pathologic analysis of the retina have demonstrated serous deposits, with compression of retinal vessel lumens, no thrombosis, and decreased adenosine diphosphatase (ADPase) activity suggesting endothelial cell dysfunction and vaso-occlusion.17 Patients do not have visual loss when the occlusions spare the macula.
Susac’s syndrome also includes sensorineural hearing loss, usually of low to mid-frequency, which can be insidious in onset, and either unilateral or bilateral.18 It is sometimes associated with ataxia, vertigo, and nausea, consistent with symptoms experienced by our patient. SG is currently awaiting audiologic evaluation to confirm physical examination findings of right-sided hearing loss.
The clinical course of Susac’s syndrome tends to be self-limited, but can recur, and varies in duration from six months to five years.16 Approximately 60% to 70% of patients experience some degree of persistent encephalopathy, which may manifest as psychiatric disturbances or dementia.6,9,16 The prognosis for recovery of hearing is variable, with some persistence of hearing loss but with improvement in tinnitus and vertigo.19 Visual loss recovery depends on the areas of the retina involved.
Initial treatment includes aggressive immunosuppression, primarily with high-dose corticosteroids and often with intravenous immunoglobulins (IVIg). Cyclophosphamide, mycophenolate mofetil, methotrexate, and azathioprine have also been used.11 After an initial pulse of corticosteroids, approximately one to three years of immunosuppression is recommended with prednisone and a steroid-sparing agent such as azathioprine.11,16 Data on use of antiplatelet agents is mixed.6,16 To date, there is one study that examines long-term outcomes in nine patients over a mean of 6.4 years.7 Corticosteroids were found to effectively treat encephalopathy with relapses as the dose was tapered. However, corticosteroids did not improve hearing loss or the development of new retinal artery occlusions.7
Our patient received methylprednisolone, 1 gram daily for five days and IVIg (400 mg/kg) daily for five days, followed by 60 milligrams of prednisone daily. Aspirin 81 milligrams daily was started. During the course of his initial treatment, he demonstrated significant neurologic improvement with decreased word-finding difficulties, steadier gait, and improved memory. His visual acuity is intact; however, he continues to suffer from right-sided hearing loss.
The Bottom Line
Susac’s syndrome is a microangiopathy characterized by encephalopathy, sensorineural hearing loss, branch retinal artery occlusions, and specific MRI findings, notably multifocal hyperintensities with involvement of the central fibers of the corpus callosum.
The differential diagnosis for patients with Susac’s syndrome is broad and includes primary angiitis of the CNS, infections, multiple sclerosis, and systemic rheumatologic diseases.
Treatment of Susac’s syndrome involves aggressive immunosuppression with high-dose corticosteroids, and depending on the severity of the manifestations, intravenous immunoglobulin and/or cyclophosphamide. Immunosuppression should be prolonged, and a steroid-sparing agent should be added, given the potential for disease relapse.
Dr. Feldman is a rheumatology fellow at Brigham and Women’s Hospital and Beth Israel Deaconess Medical Center, both in Boston. Dr. Kane-Wanger is a member of the rheumatology faculty of the Beth Israel Deaconess Medical Center.
References
- Hanley DF, Johnson RT, Whitley RJ. Yes, brain biopsy should be a prerequisite for herpes simplex encephalitis treatment. Arch Neurol. 1987;44:1289-1290.
- Steiner I, Budka H, Chaudhuri A, et al. Viral encephalitis: A review of diagnostic methods and guidelines for management. Eur J Neurol. 2005;12:331-343.
- Boesenberg C, Schulz-Schaeffer WJ, Meissner B, et al. Clinical course in young patients with sporadic Creutzfeldt-Jakob disease. Ann Neurol. 2005;58:533-543.
- Calabrese LH, Duna GF, Lie JT. Vasculitis in the central nervous system. Arthritis Rheum. 1997;40:1189-1201.
- Susac JO, Hardman JM, Selhorst JB. Microangiopathy of the brain and retina. Neurology. 1979;29:313-316.
- Susac JO. Susac’s syndrome: The triad of microangiopathy of the brain and retina with hearing loss in young women. Neurology. 1994;44:591-593.
- Aubart-Cohen F, Klein I, Alexandra JF, et al. Long-term outcome in Susac syndrome. Medicine. 2007;86:93-102.
- Nicolle MW, McLachlan RS. Microangiopathy with retinopathy, encephalopathy, and deafness (RED-M) and systemic features. Semin Arthritis Rheum. 1991;21:123-128.
- Do TH, Fisch C, Evoy F. Susac syndrome: Report of four cases and review of the literature. AJNR Am J Neuroradiol. 2004;25:382-388.
- Petty GW, Engel AG, Younge BR, et al. Retinocochleocerebral vasculopathy. Medicine. 1998;77:12-40.
- Bienfang DC, McKenna MJ, Papaliodis GN, Gonzalez RG, Stemmer-Rachamimov A. Case records of the Massachusetts General Hospital. Case 24-2011. A 36-year-old man with headache, memory loss, and confusion. N Engl J Med. 2011;365:549-559.
- Bogousslavsky J, Gaio JM, Caplan LR, et al. Encephalopathy, deafness and blindness in young women: A distinct retinocochleocerebral arteriolopathy? J Neurol Neurosurg Psychiatry. 1989;52:43-46.
- Heiskala H, Somer H, Kovanen J, Poutiainen E, Karli H, Haltia M. Microangiopathy with encephalopathy, hearing loss and retinal arteriolar occlusions: Two new cases. J Neurol Sci. 1988;86:239-250.
- Monteiro ML, Swanson RA, Coppeto JR, Cuneo RA, DeArmond SJ, Prusiner SB. A microangiopathic syndrome of encephalopathy, hearing loss, and retinal arteriolar occlusions. Neurology. 1985;35:1113-1121.
- White ML, Zhang Y, Smoker WR. Evolution of lesions in Susac syndrome at serial MR imaging with diffusion-weighted imaging and apparent diffusion coefficient values. AJNR Am J Neuroradiol. 2004;25:706-713.
- Garcia-Carrasco M, Jimenez-Hernandez C, Jimenez-Hernandez M, et al. Susac’s syndrome: An update. Autoimmun Rev. 2011;10:548-552.
- McLeod DS, Ying HS, McLeod CA, et al. Retinal and optic nerve head pathology in Susac’s syndrome. Ophthalmology. 2011;118:548-552.
- Roeser MM, Driscoll CL, Shallop JK, Gifford RH, Kasperbauer JL, Gluth MB. Susac syndrome—a report of cochlear implantation and review of otologic manifestations in twenty-three patients. Otol Neurotol. 2009;30:34-40.
- Demir MK. Case 142: Susac syndrome. Radiology. 2009;250:598-602.