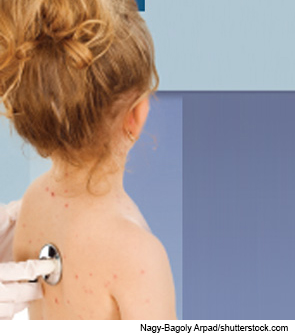
CASE 1: Recently, I (RML) received an e-mail from a 32-year-old father of a 21-month-old girl who appeared to have developed an ongoing “rash” that was identical to one he had had for the past 14 years (see Figure 1). He had seen multiple physicians and specialists, including a dermatologist, rheumatologist, allergist, ophthalmologist and otolaryngologist, but no diagnosis had been made. Seeing her with what he described as a non-itchy urticarial eruption caused him to spend the year reading extensively about diagnostic possibilities. He stumbled across Muckle‑Wells syndrome, and it seemed to explain many of his symptoms. These included episodes of arthritis beginning when he was a school-aged child, which lessened in severity as he got older. He also suffered from “migraine” headaches, which similarly lessened in severity as he grew older. He had had a non-itchy urticarial rash and bloodshot eyes (see Figure 2) from the age of 17, and at age 30 he discovered he had significant hearing loss.
His daughter had recently experienced two bouts of severe pain in her legs, and she had the same, consistent urticarial eruption that he has. She also has had an occasional bloodshot eye and a fever.
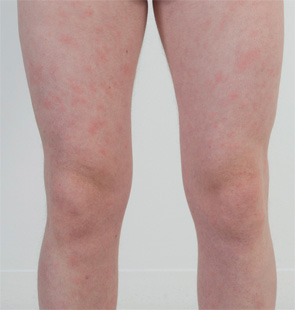
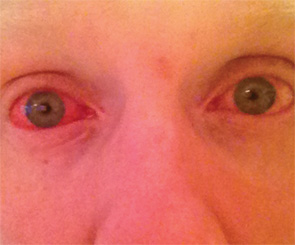
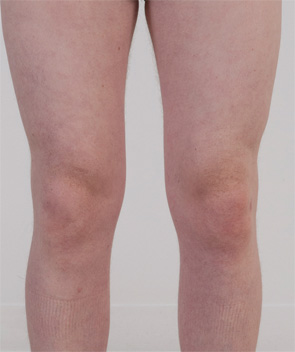
CASE 2: In 2012, a 12-year-old female of North African-Jewish origin was referred for evaluation of frequent episodes of rash, arthritis (mainly ankle), fever, “red” eyes, “migraine” headaches and mouth sores that she had experienced from the first year of her life. Acute-phase reactants were elevated, even between episodes. She had short stature and weight for her age, and was still in the pubertal developmental stage of Tanner I. She was absent from school ~80% of days. Twelve other family members (see Figure 4), including a deceased grandfather, mother, four aunts and four cousins (and since diagnosis, another two first cousins once removed), had short episodes of fever, accompanied by headache, myalgia, arthralgia and an urticarial skin rash. Prior workup found no mutations in the MEFV gene (for familial Mediterranean fever), although she was positive for the HLA-B51 antigen. The provisional diagnosis was familial Behçet’s disease, although there was no response of symptoms to colchicine and azathioprine, and only a mild improvement when corticosteroids were used.
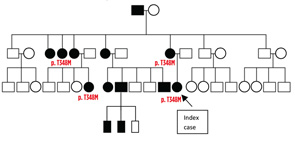
When I (PJH) first saw the patient, it was clear she had hearing difficulties, and her mother had hearing aids in both ears. The patient also had an urticarial rash and red eyes. Genetic studies discovered the T348M pathogenic variant on one allele of the NLRP3 gene in the patient, her mother and other family members. Treatment with canakinumab led to a dramatic decrease in all symptoms except headaches, and she entered a rapid growth phase, including pubertal development. There was normalization of the acute-phase reactants. Her hearing loss did not progress.
Discussion
These cases demonstrate the difficulty and delay in recognizing and diagnosing the rare autosomal-dominant cryopyrin-associated periodic syndromes (CAPS). CAPS is an autoinflammatory syndrome that consists of three classic phenotypes, described clinically over a period of 70 years, although only recently appreciated to involve similar mechanisms.
The first and mildest phenotype, described in 1940 by Kile and Rusk, was originally termed familial cold urticaria and is now called familial cold autoinflammatory syndrome (FCAS).1
The second clinical phenotype was described in 1962 and is named after the authors, Muckle-Wells syndrome (MWS).2
The third and most severe phenotype, described in the 1970s and early 1980s, is called neonatal-onset multisystem inflammatory disease (NOMID) in North America and chronic infantile neurological, cutaneous and articular syndrome (CINCA) in Europe.3
Familial occurrences of FCAS and MWS were frequently observed, and in 2001, Hal Hoffman, MD, professor of pediatrics and medicine, University of California, San Diego, and his group discovered the genetic etiology of the disease as gain of function mutations in the cold-induced autoinflammatory syndrome 1 (CIAS1) gene on the long arm of chromosome 1, encoding the cryopyrin protein.4 Later, the name of the gene was changed to the nucleotide-binding oligomerization domain-like receptor family, pyrin domain containing 3 (NLRP3) gene, based on the structure and function of the protein (also known as NLRP3). Due to the severity of the NOMID/CINCA phenotype, only rare familial cases were described, but a number of clinical features common to NOMID/CINCA and to FCAS and MWS, specifically rash, eye inflammation and hearing loss, led to the discovery by Ann-Marie Prieur, MD, from the Unit of Pediatric Immunology-Hematology and Rheumatology at the Hôpital Necker-Enfants Malades in Paris, France, and colleagues that the genetic etiology of NOMID/CINCA is identical to FCAS and MWS.5,6
Currently, it’s estimated that one in 360,000 to one in 1 million people suffer from CAPS, with the majority having MWS or an overlap phenotype.7,8 NOMID/CINCA, although dramatic, is the least common of the CAPS phenotypes, numbering perhaps several hundred patients worldwide.5 The prevalence of FCAS is not clear, but most reported patients are from the U.S.
These figures, however, may be a substantial underestimate, in part due to varied clinical presentation, lack of awareness and difficulties in genetic diagnosis. In Israel, for example, there are 26 known patients with CAPS from a population of 8 million (~1:315,000), all diagnosed since 2007 [Note: PJH, personal knowledge], with nearly 50% of them belonging to the family previously described. Data from France indicate that since 2004, steady numbers of 4–12 new genetically proven, mostly sporadic cases are discovered yearly; most familial cases were diagnosed early after the discovery of the gene responsible for CAPS.8
Clinical Manifestations & Diagnosis
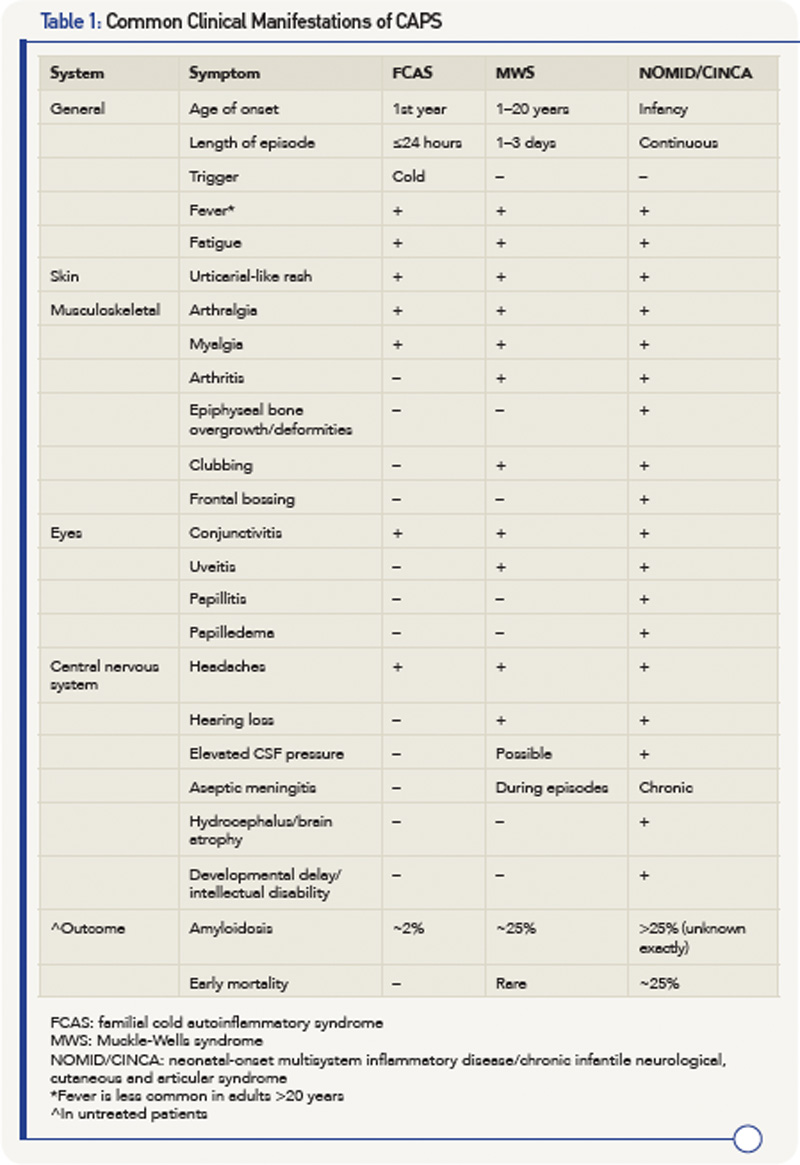
The three syndromes share many similarities, but differ in the severity of manifestations and, especially, in outcome.9 Common features include fever, urticaria-like rash, eye inflammation, musculoskeletal manifestation and headaches/central nervous system involvement. Table 1 (right) details the similarities and differences between the syndromes.
Patients may present to physicians from many specialties, including rheumatologists, pediatricians, internists, infectious disease specialists, allergists, immunologists, dermatologists, neurologists (and even neurosurgeons with hydrocephalus), ophthalmologists, otolaryngologists, orthopedists and neonatologists.10,11 In one series, 44% of patients were diagnosed with at least one other disease (as with the family in Case 2), and it took from 1.4 years (among infants born in the 21st century) to >30–40 years (adults born in the 1960s and 1970s) to be diagnosed.12,13 Prior to establishing the correct diagnosis, patients were diagnosed with—from the most frequent—conjunctivitis, other rheumatic disease, sensorineural hearing loss, urticaria and uveitis.13 Therefore, it’s extremely important that awareness of CAPS be spread to other specialists.
In potential patients, acute-phase reactants should be examined and if they are elevated without an adequate alternative explanation, CAPS (and possibly other autoinflammatory conditions) should be suspected.
There is often overlap between the three “classic” conditions, and as recently discovered, many patients may present atypically, further complicating disease recognition and diagnosis. For example, the frequency of rash in MWS varies in different series from 67–92%; thus, rash, although common and important, may not be a necessary feature in CAPS.8,12,13 One study reported significant differences in the presentation of CAPS in young children and adults, with recurrent fever and abdominal pain (i.e., inflammatory phenotype) much more frequent among children and general fatigue, musculoskeletal symptoms and hearing loss (without fever) in adults (i.e., organ-disease phenotype).12
The diagnosis is confirmed by genetic testing, with most mutations present on exon 3 of the NLRP3 gene. More than 170 gene variants have been described. In some variations/mutations, a clear phenotype-genotype correlation exists. For example, the T348M mutation is associated with more severe hearing loss (as with the family in Case 2), but many mutations are not specific for a certain phenotype, such as the wide clinical spectrum seen in families with the E311K mutation.14,15 Therefore, it’s clear that the environment and, perhaps, epigenetics influence disease severity. Further, several variants (most commonly Q703K and V198M) are found in the general population (as much as 10% for Q703K), and their clinical significance is often not clear. Many patients with these variants exhibit nonspecific inflammatory symptoms.
Unfortunately, no mutations are found in many patients when tested by traditional methods, especially with those with NOMID/CINCA (40–50%) and MWS (25–33%).9 When tested (still not widely available in commercial testing) by massively parallel sequencing, somatic mosaicism (as opposed to germline mutations in which all cell lines are affected) is detected in nearly 70% of genetically negative NOMID/CINCA patients (in 4–36% of cells, perhaps with milder neurologic sequela), but only in 12.5% of MWS patients (in 5.5–35% of cells).16,17
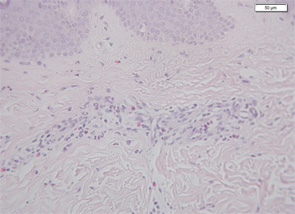
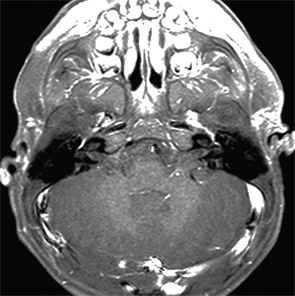
Therefore, a high level of suspicion and awareness is crucial in making the diagnosis of CAPS. There are several diagnostic aids to help diagnose these patients. These include a biopsy of the rash. Unlike classic urticaria, which is characterized by edema and the presence of mast cells, the rash in CAPS shows a dense neutrophilic infiltrate mainly in the lower (reticular) layer of the dermis, which tends to be perivascular and peri-eccrine (see Figure 5). Magnetic resonance imaging (MRI) of the brain with a focus on the inner ear often shows edema with gadolinium enhancement (occasionally also meningeal enhancement is present) (see Figure 6). Lumbar puncture during attacks may show increased opening pressure with pleocytosis and elevated protein levels. Hearing tests showing high-frequency hearing loss may be a first sign of impending deafness. A detailed eye examination may reveal uveitis in addition to conjunctivitis. In unclear cases of FCAS, placing the patient in a very cold environment for 15–20 minutes may induce an attack and rash within 30 minutes to several hours, with increased production of acute-phase reactants.
Formal diagnostic criteria for CAPS have not yet been defined. In order not to deny treatment and prevent potentially devastating damage to patients, genetic confirmation should not be a compulsory criterion. In Table 2 (above), we suggest criteria for potential clinical diagnosis. Large databases, such as the Eurofever registry, may be an aid in defining evidence-based criteria.12

Pathogenesis
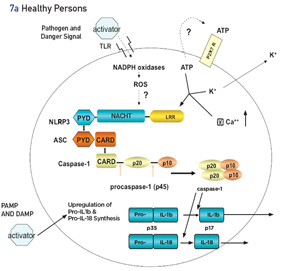
The discovery of CAPS, the mutation in the NLRP3 gene and the role of cryopyrin in the production and activity of IL-1β serve as a human laboratory that has led to a wider understanding of the innate immune system, especially that of the inflammasome and pro-inflammatory cytokines. CAPS is the prototype of IL-1β-driven autoinflammatory diseases.
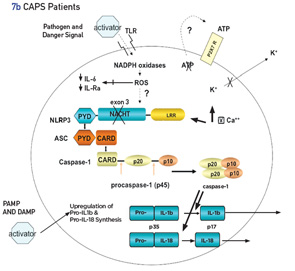
Inflammasomes are intracellular molecular complexes that respond to both external and internal agents to activate cytokines IL-1β and IL-18 (see Figures 7a and b). In health, the inflammasome is activated by many exogenous triggers, such as pathogens (via pathogen-associated molecular pattern molecules [PAMPs]) or damage-associated molecular pattern molecules (DAMPs), such as crystals (urates in gout) or cholesterol (atherosclerosis). In addition, adenosine triphosphate (ATP), as a second activating signal, is usually necessary to activate the inflammasome via an efflux of cellular potassium.18 PAMPs and DAMPs act through membrane-based, toll-like receptors and other signals, and lead to the assembly of several proteins (many with a pyrin domain), one of which is the NLRP3 inflammasome. The inflammasome activates caspase 1, which converts pro-IL-1β to active IL-1β. Other important regulators of the inflammasome are the level of intracellcular calcium (increases activity) and cyclic adenosine monophosphate (cAMP, decreases activity).19
In patients with NLRP3 mutations, which lead to cryopyrin gain of function, many of these stages are bypassed, augmented or diminished, with the net result being increased spontaneous activation of the inflammasome. In CAPS, the second signal of ATP is not necessary for activation of the inflammasome.20 In patients with CAPS, the inflammasome may be more sensitive to the calcium-sensing receptor (CASR), resulting in increased release of calcium from the endoplasmic reticulum via phospholipase C, which catalyzes inositol-1,4,5-trisphosphate production and, thereby, induces release of calcium. This leads to further spontaneous assembly and activation of the inflammasome.19 In CAPS, the binding affinity of cAMP to the mutated inflammasome is decreased, thus reducing the regulatory effect of cAMP on its activity.19
In addition to IL-1β, other cytokines and even the adaptive immune system may contribute to inflammation in CAPS patients, although the effectiveness of treatment with IL-1 inhibitors points toward the predominance (or perhaps upstream) effect of IL-1 in CAPS. IL-17 production is induced in CAPS patients by IL-1, resulting in increased TH-17 cells in CAPS patients.21 These effects are reversed by IL-1β inhibition. IL-18 is markedly increased in CAPS patients. In mouse models of CAPS, IL-18 appears to play an important role in the early phase of disease.22 In these models, IL-1 inhibition may not result in clinical improvement in young mice. Hence, in the rare patient who does not respond well to IL-1 inhibition, particularly at a very early age, IL-18 may play a more important role in the pathogenesis of disease. Evidence for phagocyte activation in CAPS patients is shown by elevated levels of the phagocyte-specific molecules myeloid-related protein (MRP) 8 and MRP14 in untreated patients, with a marked decrease (although not always to normal levels) with IL-1 inhibitor treatment.23 Oxidative stress of monocytes has been shown to lead to a decrease in IL-6 and IL-1 receptor antagonist in CAPS patients, exacerbating IL-1 overproduction.24 Defective cell death mechanism related to cryopyrin mutations has been shown to be associated with tissue infiltration of neutrophils.25
The importance of discovering the cause and pathogenesis of CAPS has wider implications than just the diagnosis and treatment of patients with this rare condition. Through the knowledge gained by this real-life model of the innate immune system, the role of the inflammasome and IL-1 in causing disease has led to discoveries with profound impact on common conditions. For example, gout attacks have been shown to be mediated by uric acid crystals triggering activation of the NLRP3 inflammasome.26 IL-1 inhibition is effective in treating and preventing gout attacks.27 Metabolic syndromes, such as type 2 diabetes, cause an increase in inflammasome activation of IL-1β, and coronary artery inflammation, as well as postmyocardial infarction damage is, at least in part, mediated via IL-1.27
Treatment
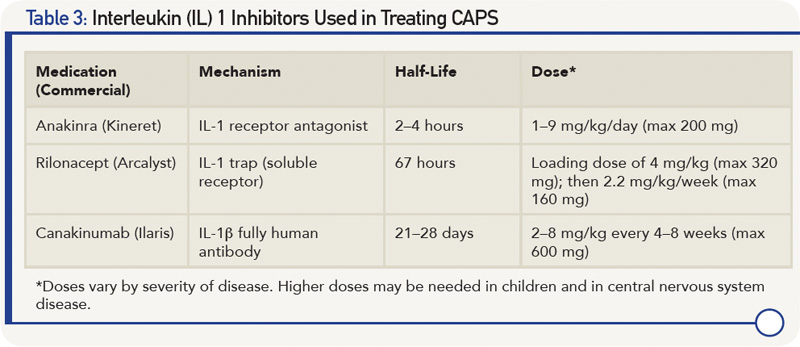
As further proof of concept that CAPS is an IL-1-driven disease is the dramatic clinical and laboratory response to all IL-1 inhibitors studied thus far (see Table 3) with continued long-term response to therapy.28-31 IL-1 inhibitors not only prevent IL-1β from binding to the IL-1 receptor, thus reducing cell signaling, but also decrease IL-1 production to levels in healthy subjects.32 In the controlled studies of rilonacept and canakinumab, >85% of patients (primarily Muckle-Wells phenotype) were complete responders, with the response occurring in the majority of patients within one week of starting therapy.28,29 In the anakinra study of NOMID/CINCA, discontinuing anakinra resulted in a flare after a median of 5 days. As a result, the withdrawal phase of the study protocol was discontinued for ethical reasons after 11 of the 18 participants had been withdrawn from treatment.30
There are several important treatment issues in CAPS:
- The most important issue is that existing damage is usually not reversible; thus, diagnosis and initiation of treatment need to be prior to development of damage. IL-1 inhibition improves existing hearing loss in only ~20–33% of patients and not after deafness has already occurred.14 Treatment usually prevents further hearing deterioration.
- Bone deformities in NOMID/CINCA are not reversible.33 Rather than being mediated by IL-1, bone overgrowth in NOMID/CINCA is mediated by osteoblast progenitor cells that form fibroblastoid tumors. These cells are stimulated by cAMP-dependent protein kinase A through activation of caspase-1 leading to over-expression of the early osteoblast factor Ets-1.34 IL-1 inhibition also does not prevent formation of these deformities.33
- In many patients, the dose and frequency of IL-1 inhibition therapy need adjustment over time, particularly in children and in those with a more severe phenotype. Anakinra can be increased up to 8–9 mg/kg/day and canakinumab to 600 mg (in children ≤40 kg, 8 mg/kg) every four weeks.31,33,35 Other patients may need less frequent administration of canakinumab.
- It’s still unclear whether one type of IL-1 inhibition is better than others in treating central nervous system disease, particularly in patients with NOMID/CINCA, because it’s still not known whether there is adequate penetration of canakinumab across the blood–brain barrier to achieve adequate CNS penetration. In human and mouse experimental models, anakinra has been shown to have good brain penetration, but data are lacking for other IL-1 inhibitors.36
- It’s still unclear whether it’s necessary to use IL-1 inhibitors in patients with a pure FCAS phenotype, without a family history of hearing loss or amyloidosis. For untreated FCAS patients, close monitoring of their hearing and urinalysis (for proteinuria as a first sign of amyloidosis) is warranted.
- Although clinical and laboratory remission is common, many patients continue to show immunologic disease activity with continued elevated innate immunity gene expression with high levels of MRP8 and 14 and IL-18.21
Summary
Although rare, it’s likely that CAPS is underdiagnosed, in particular the FCAS and MWS phenotypes. Greater awareness of the varied clinical manifestations spread across many medical specialties is essential for early diagnosis. Early diagnosis and treatment are essential for prevention of irreversible damage, in addition to improvement in the quality of life of these patients. The lessons taught to us by this rare disease, which is the prototype of IL-1β-driven autoinflammatory diseases, have implications on understanding the innate immune system and on the pathogenesis and, perhaps, treatment of many common conditions.
Philip J. Hashkes, MD, MSc, works in the Pediatric Rheumatology Unit of Shaare Zedek Medical Center, in Jerusalem, Israel.
Ronald M. Laxer, MDCM, FRCPC, works in the Division of Rheumatology, in the Departments of Pediatrics and Medicine, The Hospital for Sick Children and University of Toronto, in Ontario, Canada.
References
- Kile RL, Rusk HA. A case of cold urticaria with an unusual family history. JAMA. 1940;114(12):1067–1068.
- Muckle TJ, Wells M. Urticaria, deafness and amyloidosis: A new heredo-familial syndrome. QJ Med. 1962 Apr;31:235–248.
- Prieur AM, Griscelli C. Arthropathy with rash, chronic meningitis, eye lesions and mental retardation. J Pediatr. 1981 Jul;99(1):79–83.
- Hoffman HM, Mueller JL, Brodie DH, et al. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001 Nov;29(3):301–305.
- Hashkes PJ, Lovell DJ. Recognition of infantile-onset multisystem inflammatory disease as a unique entity. J Pediatr. 1997 Apr;130(4):513–515.
- Feldmann J, Prieur AM, Quartier P, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet. 2002 Jul;71(1):198–203.
- Lainka E, Neudorf U, Lohse P, et al. Analysis of cryopyrin-associated periodic syndromes (CAPS) in German children: Epidemiological, clinical and genetic characteristics. Klin Padiatr. 2010 Nov;222(6):356–361.
- Cuisset L, Jeru I, Dumont B, et al. Mutations in the autoinflammatory cryopyrin-associated periodic syndrome gene: Epidemiological study and lessons from eight years of genetic analysis in France. Ann Rheum Dis. 2011 Mar;70(3):495–499.
- Aksentijevich I, D Putnam C, Remmers EF, et al. The clinical continuum of cryopyrinopathies: Novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 2007 Apr;56(4):1273–1285.
- Krause K, Grattan CE, Bindslev-Jensen C, et al. How not to miss autoinflammatory diseases masquerading as urticaria. Allergy. 2012 Dec;67(12):1465–1474.
- Kitley JL, Lachmann HJ, Pinto A, Ginsberg L. Neurologic manifestations of the cryopyrin-associated periodic syndrome. Neurology. 2010 Apr;74(16):1267–1270.
- Toplak N, Frenkel J, Ozen S, et al. An international registry on autoinflammatory diseases: The Eurofever experience. Ann Rheum Dis. 2012 Jul;71(7):1177–1182.
- Kuemmerle-Deschner JB, Dembi Samba S, Tyrrell PN, et al. Challenges in diagnosing Muckle-Wells syndrome: Identifying two distinct phenotypes. Arthritis Care Res (Hoboken). 2014 May;66(5):765–772.
- Kuemmerle-Deschner JB, Koitschev A, Ummenhofer K, et al. Hearing loss in Muckle-Wells syndrome. Arthritis Rheum. 2013 Mar;65(3):824–831.
- Kuemmerle-Deschner JB, Lohse P, Koetter I, et al. NLRP3 E311K mutation in a large family with Muckle-Wells syndrome—Description of a heterogeneous phenotype and response to treatment. Arthritis Res Ther. 2011;13(6):R196.
- Tanaka N, Izawa K, Saito MK, et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: Results of an International Multicenter Collaborative Study. Arthritis Rheum. 2011 Nov;63(11):3625–3632.
- Nakagawa K, Gonzalez-Roca E, Souto A, et al. Somatic NLRP3 mosaicism in Muckle-Wells syndrome. A genetic mechanism shared by different phenotypes of cryopyrin-associated periodic syndromes. Ann Rheum Dis. 2013 Dec 10; [Epub ahead of print].
- Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006 Mar 9;440(7081):228–232.
- Lee GS, Subramanian N, Kim AI, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012 Dec 6;492(7427):123–127.
- Gattorno M, Tassi S, Carta S, et al. Pattern of interleukin-1beta secretion in response to lipopolysaccharide and ATP before and after interleukin-1 blockade in patients with CIAS1 mutations. Arthritis Rheum. 2007 Sep;56(9):3138–3148.
- Lasigliè D, Traggiai E, Federici S, et al. Role of IL-1 beta in the development of human T(H)17 cells: Lesson from NLPR3 mutated patients. PLoS One. 2011;6(5):e20014.
- Brydges SD, Broderick L, McGeough MD, et al. Divergence of IL-1, IL-18, and cell death in NLRP3 inflammasomopathies. J Clin Invest. 2013 Nov 1;123(11):4695–4705.
- Wittkowski H, Kuemmerle-Deschner JB, Austermann J, et al. MRP8 and MRP14, phagocyte-specific danger signals, are sensitive biomarkers of disease activity in cryopyrin-associated periodic syndromes. Ann Rheum Dis. 2011 Dec;70(12):2075–2081.
- Carta S, Tassi S, Delfino L, et al. Deficient production of IL-1 receptor antagonist and IL-6 coupled to oxidative stress in cryopyrin-associated periodic syndrome monocytes. Ann Rheum Dis. 2012 Sep;71(9):1577–1581.
- Satoh T, Kambe N, Matsue H. NLRP3 activation induces ASC-dependent programmed necrotic cell death, which leads to neutrophilic inflammation. Cell Death Dis. 2013 May 23;4:e644.
- Martinon F, Petrilli V, Mayor A, et al. Gout associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006 Mar 9;440(7081):237–241.
- Dinarello CA. Blocking interleukin-1β in acute and chronic autoinflammatory diseases. J Intern Med. 2011 Jan;269(1):16–28.
- Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (interleukin-1 trap) in patients with cryopyrin-associated periodic syndromes. Arthritis Rheum. 2008 Aug;58(8):2443–2452.
- Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, et al, for the Canakinumab in CAPS Study Group. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med. 2009 Jun 4;360(23):2416–2425.
- Goldbach-Mansky R, Dailey NJ, Canna SW, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1 beta inhibition. N Engl J Med. 2006 Aug 10;355(6):581–592.
- Kuemmerle-Deschner JB, Hachulla E, Cartwright R, et al. Two-year results from an open-label, multicentre, phase III study evaluating the safety and efficacy of canakinumab in patients with cryopyrin-associated periodic syndrome across different severity phenotypes. Ann Rheum Dis. 2011 Dec;70(12):2095–2102.
- Lachmann HJ, Lowe P, Felix SD, et al. In vivo regulation of interleukin 1beta in patients with cryopyrin-associated periodic syndromes. J Exp Med. 2009 May 11;206(5):1029–1036.
- Sibley CH, Plass N, Snow J, et al. Sustained response and prevention of damage progression in patients with neonatal-onset multisystem inflammatory disease treated with anakinra: A cohort study to determine three- and five-year outcomes. Arthritis Rheum. 2012 Jul;64(7):2375–2386.
- Almeida MQ, Tsang KM, Cheadle C, et al. Protein kinase A regulates caspase-1 via Ets-1 in bone stromal cell-derived lesions: A link between cyclic AMP and pro-inflammatory pathways in osteoblast progenitors. Hum Mol Genet. 2011 Jan 1;20(1):165–175.
- Caorsi R, Lepore L, Zulian F, et al. The schedule of administration of canakinumab in cryopyrin associated periodic syndrome is driven by the phenotype severity rather than the age. Arthritis Res Ther. 2013 Feb 26;15(1):R33.
- Galea J, Ogungbenro K, Hulme S, et al. Intravenous anakinra can achieve experimentally effective concentrations in the central nervous system within a therapeutic time window: Results of a dose-ranging study. J Cereb Blood Flow Metab. 2011 Feb;31(2):439–447.

