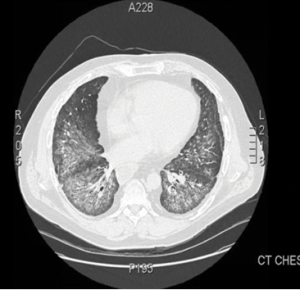
Figure 1.
This computed tomography of the chest shows bilateral, diffuse, ground-glass opacities and nodular opacities with intralobular septal thickening.
Antisynthetase syndrome (AS) is strongly associated with the presence of antibodies to aminoacyl-transfer RNA (tRNA) synthetases (ARSs) that are implicated in the pathogenesis of myositis and interstitial lung disease (ILD).
Antibodies against eight antisynthetases have been identified and are detected in 16–26% of patients with idiopathic inflammatory myopathies (IIM).1 Serum assays for five of these antibodies are commercially available: anti-histidyl (Jo-1), anti-threonyl (PL-7), anti-alanyl (PL-12), anti-isoleucyl (OJ) and anti-glycl (EJ). Anti-Jo-1 antibodies are seen more commonly in patients with polymyositis and dermatomyositis compared with the others that are found less frequently.2 These antibodies usually target the enzymatic component of tRNA synthetases; only anti-PL-12 recognizes tRNA directly.
The physical examination revealed decreased bilateral breath sounds, puffy fingers, hyperkeratotic skin over the palmar surface of both hands, no clinical evidence of synovitis and normal muscle strength.
Laboratory analysis revealed mild inflammation with C-reactive protein (CRP) of 15 mg/L, normal creatine phosphokinase (CPK) and normal liver function tests. Antinuclear antibodies (ANA) were negative, and no anti-cytoplasmic staining was detected on indirect immunofluorescence. Rheumatoid factor and anticyclic citrullinated protein antibodies were negative.
An additional workup showed negative anti-neutrophil cytoplasmic antibodies, negative anti-SSA antibodies, and negative PM-Scl antibodies. Pulmonary function tests revealed mild restrictive ventilator defect with severe reduction in diffusion capacity and forced vital capacity (FVC). Forced expiratory volume in 1 second (FEV 1) was 75%, and carbon monoxide diffusion capacity (DLCO) was 36%. High-resolution computed tomography (CT) of the chest demonstrated bilateral pulmonary infiltrates with alveolitis.
The patient was monitored regularly with clinical, laboratory, radiological and lung function evaluations for four years. He developed progression of ILD with declining pulmonary function tests. High-resolution CT imaging showed bilateral pulmonary infiltrates and alveolitis with diffuse, bilateral, ground-glass opacities and nodular infiltrates with intraseptal wall thickening compatible with nonspecific interstitial pneumonia (see Figure 1).
The patient was refractory to conventional immunosuppressive therapies including systemic corticosteroids, cyclophosphamide, mycophenolate mofetil, azathioprine, cyclosporine and intravenous immune globulin. He required multiple hospitalizations for acute respiratory failure requiring intubation, and he developed recurrent infections for which immunosuppressive therapy had to be discontinued.
In February 2013, the patient’s pulmonary status declined and his supplemental oxygen requirements increased. Physical examination showed 4.5/5 strength on hip flexors, 5/5 in all remaining major flexors and extensors with normal grip. The patient had hyperkeratotic skin lesions on his hands consistent with mechanic’s hands. Pulmonary function tests showed total lung capacity to be 62% of predicted. Forced vital capacity was 60% of predicted. DLCO was 29%, and FEV-1 was normal.
A CT of his chest demonstrated ground-glass opacities and honeycombing. An echocardiogram showed normal ejection fraction and diastolic function. His pulmonary artery systolic pressure was 25 mmHg.
Laboratory results included an erythrocyte sedimentation rate of 86 mm/h, CRP 181.0 mg/L and an elevated serum CPK level (1,500 U/L, normal ≤145 U/L).
The clinical picture was compatible with AS, which prompted the search for antisynthetase antibodies. Anti-Jo-1 antibodies were negative, but more specific testing (ImmunoDOT technique) revealed the presence of anti-PL-12 antibodies. To treat the ILD that had been refractory to all other immunosuppressive therapies, he was given two doses of 1,000 mg rituximab every two weeks, with the second dose given in November 2016.
He improved and had stable pulmonary function tests at his six-month outpatient follow-up appointment with our clinic.
This case emphasizes the need to consider AS in patients with ILD & undifferentiated connective tissue disease and to screen for ARS antibodies, particularly in the presence of typical clinical features (mechanic’s hands), a negative ANA & a positive anti-cytoplasmic signal.
Discussion
AS is a rare, chronic autoimmune disease of unknown etiology. It belongs to the subgroup of idiopathic inflammatory muscle diseases. It is a clinical entity characterized by interstitial lung disease (ILD), inflammatory myopathy, arthritis, Raynaud’s phenomenon, fever, mechanic’s hands and the presence of antisynthetase antibodies.
A diagnostic feature is the presence of serum autoantibodies that recognize the ARS antibodies. Antisynthetase antibodies are ARS antibodies that are directed against the enzymes that attach dedicated amino acids to their cognate tRNA in the process of polypeptide synthesis.
Theoretically, ARS antibodies could be targeting synthetases for all 20 existing amino acids. However, to date, only eight ARS antibodies have been identified, of which anti-Jo-1 is the most prevalent, comprising two-thirds of identified ARS antibodies.3 Others are less frequent and include anti-PL-12, anti-PL-7, anti-OJ, anti-EJ, anti-asparaginyl-tRNA synthetase, anti-phenylalanyl-tRNA synthetase and anti-tyrosyl tRNA synthetase.4
The high prevalence of ILD in patients carrying ARS antibodies and the need for specific immunosuppressive treatment emphasize the need for timely detection of this clinical syndrome. This is, however, often severely hampered by the variable time relationship between the onset of ILD and the onset of myositis or other ASS-specific features. ILD can precede (10–30%), occur concurrently (53–70%) or follow (6–20%) the onset of myositis.5,6
Organized multidisciplinary follow-up by both pulmonologists and rheumatologists increases the alertness for subtle clinical signs suggesting an underlying AS.
When faced with a patient with AS, the identification of the underlying ARS antibody specificity is relevant. Increasing recent evidence supports clinical heterogeneity between anti-Jo-1- and non-Jo-1-positive patients with differences in presenting symptoms and prognosis. On the one hand, patients positive for anti-Jo-1 frequently present with muscle and joint involvement and severe and treatment-resistant myositis. On the other hand, non-Jo-1-positive patients often present with nonmyositis connective tissue disease symptoms (Raynaud’s, cutaneous changes, and constitutional symptoms) and pulmonary manifestations.6-8
Anti-PL-7- and anti-PL-12-positive patients cluster together with a 98% prevalence of ILD and isolated ILD in 62%. The behavior of anti-PL-7- and anti-PL-12-positive patients is substantially different from anti-Jo-1-positive patients (ILD in 78%).9
Identification of the ARS antibody allows for prognostic stratification. Recent reports indicate that non-Jo-1-positive patients have decreased survival compared with anti-Jo-1-positive patients (five- and 10-year unadjusted survival of 90% and 70% in anti-Jo-1-positive patients, and 75% and 47% in non-Jo-1-positive patients).8

(click for larger image)
Table 1: Myositis-Specific Autoantibodies
In one study that compared the characteristics between AS patients with anti-Jo-1 and those with anti-PL-7/PL-12 antibodies, the anti-Jo-1-positive patients more often developed severe myositis and joint manifestations, including erosive arthritis and often exhibited cancer, whereas anti-PL-7/PL-12-positive patients more commonly experienced early and severe ILD.10
Anti-PL-7/PL-12-positive patients more frequently had gastrointestinal complications, especially intestinal pseudo-obstruction. A retrospective study of 17 cases to examine the clinical manifestations of AS positive for anti-PL-12 antibodies showed that all patients presented with ILD, and mild myositis was seen in 41% of the patients.11 Raynaud’s phenomenon and general impairment were common, whereas rheumatic and dermatological symptoms were uncommon. A high prevalence of esophageal involvement (23.5%) was seen. AS appeared as an isolated diffuse interstitial pneumonia in 29.4% of patients.
The presence of anti-PL-12 antibodies in patients with presumed ILD may constitute a subset of patients who may have a favorable response to treatment with improved prognosis of their lung disease.12 However, we do not have any longitudinal data to characterize disease behavior in these patients. There is a need for prospective studies to provide further insight into the clinical consequences of anti-PL-12 antibodies and idiopathic ILD.
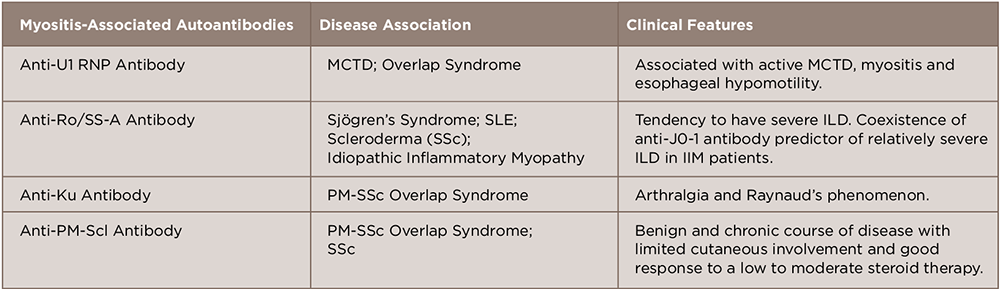
(click for larger image)
Table 2: Myositis-Associated Autoantibodies
This case emphasizes the need to consider AS in patients with ILD and undifferentiated connective tissue disease and to screen for ARS antibodies, particularly in the presence of typical clinical features (mechanic’s hands), a negative ANA and a positive anti-cytoplasmic signal. The severity of anti-PL-12 AS varies because of the constant pulmonary involvement. ILD is the predominant prognosis factor and is notable in cases associated with pulmonary hypertension.
Myositis-Related Autoantibodies
During the past decade, novel myositis-specific and myositis-associated autoantibodies have been identified that help us in the clinical diagnosis, classification, prediction of prognosis and choice of treatment in patients with idiopathic inflammatory myopathy (IIM).13
A number of antibodies detected in this disease can be subcategorized as myositis-specific and myositis-associated autoantibodies.14 These are closely associated with the clinical manifestations of polymyositis/dermatomyositis, such as symptoms, complications, reactivity to therapy and prognosis. Although considerable progress has been made in elucidating the association between genotype, serotype and clinical phenotype, further characterization of autoantigens and autoantibodies will help us understand the pathophysiology and provide us with a new therapeutic strategy in treating IIM (see Tables 1 and 2).
Conclusion
In conclusion, anti-PL-12-positive patients have a prevalence of ILD of 90–100%, with a high prevalence of isolated ILD at presentation, a lower frequency of myositis with a better response to therapy and an overall poorer prognosis compared with anti-Jo-1-positive patients.
 Quretul Quresh, MD, is a rheumatology fellow at Ochsner Medical Center, New Orleans. She completed her internal medicine residency at the University of Massachusetts School of Medicine, Berkshire Medical Center, Pittsfield, Mass. Her clinical and research interests are in the role of T cell homeostasis in rheumatoid arthritis and pathogenesis of PD1 pathway in inflammatory arthritis.
Quretul Quresh, MD, is a rheumatology fellow at Ochsner Medical Center, New Orleans. She completed her internal medicine residency at the University of Massachusetts School of Medicine, Berkshire Medical Center, Pittsfield, Mass. Her clinical and research interests are in the role of T cell homeostasis in rheumatoid arthritis and pathogenesis of PD1 pathway in inflammatory arthritis.
 Stephen Lindsey, MD, is a clinical professor of medicine at Louisiana State University (LSU) School of Medicine in New Orleans and a rheumatologist at Ochsner Medical Center. He is a graduate of LSU Medical School, did his internal medicine residency at Letterman Army Medical Center, San Francisco, and completed a rheumatology fellowship at Walter Reed Army Medical Center, Washington, D.C. His research interests include osteoporosis and the role of monoclonal antibodies and JAK inhibitors in rheumatoid arthritis.
Stephen Lindsey, MD, is a clinical professor of medicine at Louisiana State University (LSU) School of Medicine in New Orleans and a rheumatologist at Ochsner Medical Center. He is a graduate of LSU Medical School, did his internal medicine residency at Letterman Army Medical Center, San Francisco, and completed a rheumatology fellowship at Walter Reed Army Medical Center, Washington, D.C. His research interests include osteoporosis and the role of monoclonal antibodies and JAK inhibitors in rheumatoid arthritis.
References
- Kalluri M, Sahn SA, Oddis CV, et al. Clinical profile of Anti-PL2 autoantibody. Cohort study and review of the literature. Chest. 2009 Jun;135(6):1550–1556.
- Targoff IN. Autoantibodies and their significance in myositis. Curr Rheumatol Rep. 2008 Aug;10(4):333–340.
- Hervier B, Benveniste O. Clinical heterogeneity and outcomes of antisynthetase syndrome. Curr Rheumatol Rep. 2013 Aug;15(8):349.
- Mahler M, Miller FW, Fritzler MJ. Idiopathic inflammatory myopathies and the anti-synthetase syndrome: A comprehensive review. Autoimmune Rev. 2014 Apr–May;13(4–5):367–371.
- Labirua-Iturburu A, Selva-O’Callaghan A, Vincze M, Danko K, et al. Anti-PL-7 (anti-threonyl-tRNA synthetase) antisynthetase syndrome: Clinical manifestations in a series of patients from a European multicenter study (EUMYONET) and review of the literature. Medicine (Baltimore). 2012 Jul;91(4):206–211.
- Marie I, Josse S, Decaux O, et al. Clinical manifestations and outcome of anti-PL7 positive patients with antisynthetase syndrome. Eur J Intern Med. 2013 Jul;24(5):474–479.
- Lega JC, Fabienn N, Reynaud Q, et al. The clinical phenotype associated with myositis-specific and associated autoantibodies: A meta-analysis revisiting the so-called antisynthetase syndrome. Autoimmune Rev. 2014 Sep;13(9):883–891.
- Aggarwal R, Cassidy E, Fertig N, et al. Patients with non-Jo-1 anti-tRNA-synthetase autoantibodies have worse survival than Jo-1 positive patients. Ann Rheum Dis. 2014 Jan;73(1):227–232.
- Hervier B, Devilliers H, Stanciu R, et al. Hierarchical cluster and survival analyses of antisynthetase syndrome: Phenotype and outcome are correlated with anti-tRNA synthetase antibody specificity. Autoimmune Rev. 2012 Dec;12(2):210–217.
- Marie I, Josse S, Decaux O, et al. Comparison of long-term outcome between anti-Jo1- and anti-PL7/PL12 positive patients with antisynthetase syndrome. Autoimmune Rev. 2012 Aug;11(10):739–745.
- Hervier B, Wallaert B, Hachulla E, et al. Clinical manifestations of anti-synthetase syndrome positive for anti-alanyl-tRNA synthetase (anti-PL12) antibodies: A retrospective study of 17 cases. Rheumatology (Oxford). 2010 May;49(5):972–976.
- Yoshifuji H, Fuji T, Kobayashi S, et al. Anti-aminoacyl tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity. 2006 May;39(3):233–253.
- Targoff IN. Immune manifestations of inflammatory muscle disease. Rheum Dis Clin North Am. 1994 Nov;20(4):857–880.
- Nakashima R, Mimori T. Clinical and pathophysiological significance of myositis-specific and myositis-associated autoantibodies. Int J Clin Rheumatol. 2010;5(5):523–536.

