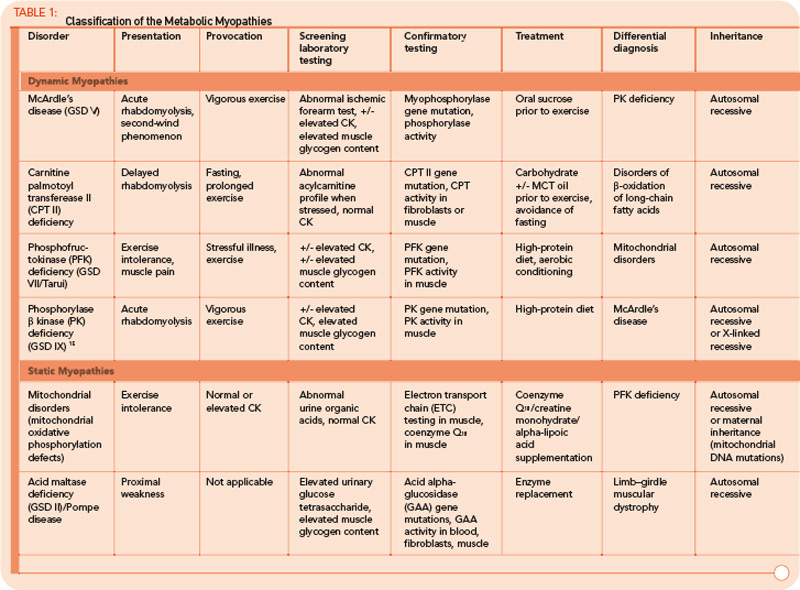
FAO Disorders
FAO (b-oxidation of fatty acids) is the major source of energy during periods of sustained, low-intensity exercise or prolonged fasting. In infants, these disorders typically present during periods of illness or poor oral intake. In children and adults, exercise intolerance and myoglobinuria are the most common presenting features. The major disorders of lipid metabolism that present with isolated myopathy include: 1) carnitine palmitoyltransferase II (CPT II) deficiency, 2) long-chain acyl-CoA dehydrogenase (LCHAD), 3) trifunctional protein including LCHAD, and 4) very long-chain acyl-CoA dehydrogenase (VLCAD) defects.10 Of these, CPT II deficiency is the most prevalent and is the most common overall cause of hereditary recurrent myoglobinuria.
Case 2
A 69-year-old female presented to the metabolic clinic following admission through the emergency room after a day of moving boxes on a cold December day. Symptoms included muscle pain accompanied by very dark colored urine. Physical examination was noncontributory. Initial laboratory evaluation revealed serum CK elevated to 32,000, elevated hepatic transaminases, and myoglobinuria. Pertinent medical history included initiation of atorvastatin therapy one week earlier for hypercholesterolemia, absence of previous episodes of muscle pain, and hypothyroidism treated with levothyroxine. Subsequently, atorvastatin was discontinued. Family history was negative for exercise intolerance or other neuromuscular symptoms. Laboratory testing obtained at the initial clinic visit revealed normal serum CK, plasma lactate, plasma carnitine, and plasma acylcarnitine profile. CPT II gene sequencing revealed a single inactivating mutation, which implied either carrier or affected status. Enzyme testing of cultured fibroblasts demonstrated a deficiency of carnitine palmitoyltransferase II, and the patient was advised to avoid prolonged fasting and to ingest beverages containing carbohydrates prior to and during any significant exercise. She experienced two episodes of rhabdomyolysis requiring hospitalization over the next three years. In contrast with the earlier normal acylcarnitine profile, subsequent acylcarnitine profiles revealed elevated long-chain acylcarnitines in a pattern consistent with CPT II deficiency (C16, C18:1, and C18:2).
CPT II Deficiency
CPT II is a protein located on the inner surface of the inner mitochondrial membrane and serves a critical role in the conversion of long-chain acylcarnitines back into long-chain acyl-CoA species. Long-chain acyl-CoA species subsequently undergo b-oxidation, which provides acetyl-CoA to the tricarboxylic acid cycle, driving synthesis of ATP via the electron transport chain.


