In 1994, the first human gene that resembled Toll was identified by Nobuo Nomura and colleagues, and in 1997, Ruslan Medshitov and Charles Janeway identified that the human Toll receptor was capable of activating NF-kB and inducing proinflammatory cytokines, which we know are important in the pathogenesis of RA. The product of the human Toll gene has subsequently come to be known as the TLR. In 1998, the group, led by Bruce Beutler, who also was awarded a Nobel Prize in 2011, identified that lipopolysaccharide (LPS) from gram-negative, bacteria-activated TLR4. Subsequently, a total of 10 human TLRs have been characterized.
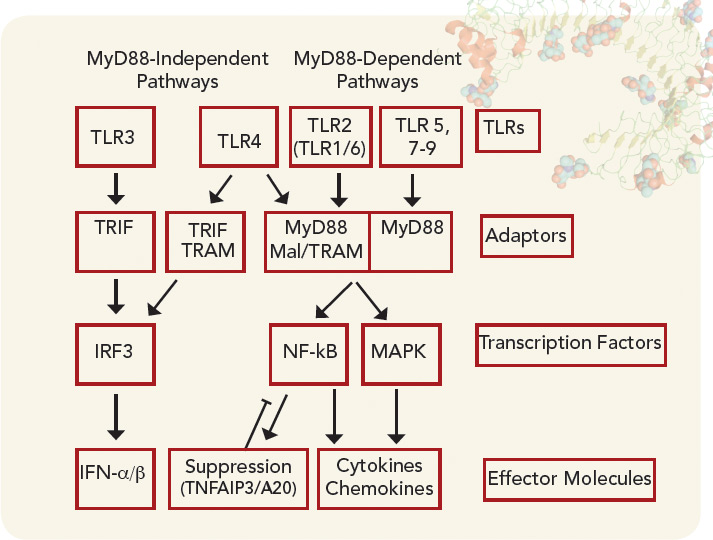
The function of TLRs can be separated based on the location of their expression within the cell either extracellularly or within the endosome (see Figure 1). Although TLRs are critical for the function of monocytes, macrophages, dendritic cells, and natural killer cells, they are also expressed by other cell types, such as fibroblasts and endothelial cells. Ligation of TLRs results in the activation of NF-kB and mitogen-activated protein kinases including Jun N-terminal Kinase, p38, and extracellular signal-regulated kinase, which promote the expression of proinflammatory cytokines, chemokines, and matrix metalloproteinases. TLRs expressed on antigen presenting cells such as macrophages and dendritic cells contribute to the induction of adaptive immunity by T and B lymphocytes by contributing to the maturation of dendritic cells to promote efficient antigen presentation and by the release of cytokines such as interferons, interleukin (IL) 1β, and IL-6, which promote lymphocyte differentiation and activation. The induction of autoantibodies in RA, including rheumatoid factor and antibodies to citrullinated proteins (ACPAs) have been shown to require antigen-presenting cells, suggesting that TLRs are involved in the initiation of autoimmunity in RA.2 How TLRs might contribute to the persistence or progression of RA will be discussed below.
Genetic Background
The genetic background associated with RA first came to light in 1976, when Peter Stastny performed allogeneic mixed lymphocyte cultures and found that the cells from patients with RA were less likely to activate cells from other patients with RA than were cells from unrelated individuals without RA.3 This suggested that there were similarities in the major histocompatibility complex (MHC) region that were shared by patients with RA. These studies led to the identification that HLA-DRB1 alleles, which include shared epitopes (SEs), are associated with RA.4 The SEs are enriched in positively charged arginines around the antigen-binding grove of the third hypervariable region of a subset of HLA-DRB1 alleles that included *0401, *04014, and *0101. A SE is present in about 60% to 70% of patients with RA. More recent studies have identified that the SE is more strongly associated with ACPAs than with the presence of RA itself.5 A mechanism contributing to the production of ACPAs by SE-positive individuals appears to be related to the increased avidity of binding of citrullinated peptides to the SE. Citrullination is a posttranslational modification mediated by peptidyl arginine deiminase, which converts the positively charged amino group on arginine to a neutral oxygen. This increased binding appears to result in the enhanced ability of citrullinated peptides to bind to SEs on antigen-presenting cells, resulting in the activation of T and B lymphocytes and in the induction of ACPAs. However, the presence of SE alleles is not uncommon, detected in almost 30% of the Caucasian population of European descent and 60% of the patients with RA.6
Although ACPAs are strongly associated with RA, the vast majority of individuals who possess the SE do not develop RA. Genome-wide association studies employing single-nucleotide polymorphisms (SNPs) have identified more than 30 different additional non-MHC genetic associations in patients with RA. The majority of the associations have been identified in ACPA-positive patients, although the PTPN22, CTLR4, and IL2RB SNPs have also been associated with those who are ACPA negative, in some but not all studies, and they appear to contribute less overall risk in the ACPA-negative patients.
The functions of the genes associated with RA, other than the MHC locus, are just beginning to be understood. SNPs in PTPN22 have been associated with a number of antibody-mediated autoimmune diseases, including RA and systemic lupus erythematosus.7 PTPN22, which is expressed primarily in hematopoietic cells, is a phosphatase that is capable of inactivating certain aspects of cell activation. Recent studies have observed that individuals expressing the PTPN22 allele encoding a R620W variant that is associated with autoimmune diseases demonstrated deficiencies in the deletion of autoreactive B cells, which may contribute to increased autoantibody production.8 Further expression of the disease-associated variant in mice resulted in enhanced activation of lymphocytes and dendritic cells, which may increase autoantibody production. Of great interest, the disease associated variant of PTPN22 containing the R620W, when expressed in experimental mice, was rapidly degraded compared with the other variants.7 Supporting the relevance of more rapid degradation to disease, both healthy controls and patients with RA who carried two copies of the risk allele demonstrated reduced levels of PTPN22 in peripheral blood mononuclear cells. Together, these studies suggest that the disease-associated variant of PTPN22 results in more rapid degradation of the phosphatase, leading to an increase in lymphocyte and dendritic cell activation, permitting the survival of autoreactive B cells.
SNPs in TNFAIP3 or A20 gene are also associated with RA. TNFAIP3 is generally weakly expressed, but is strongly and rapidly induced by activation through TLRs or TNF receptors, mediated through NF-kB activation. The induced TNFAIP3 suppresses ongoing activation resulting from TLR ligation, providing an important mechanism to attenuate the inflammatory response. Of great relevance to RA, mice deficient in TNFAIP3, specifically in macrophages, develop a spontaneous arthritis resembling RA.9 However, it is not clear how the RA-associated TNFAIP3 may function. Therefore, it is possible that the SE plus PTPN22 and TNFAIP3 SNPs, as well as any of the other identified SNPs, in combination, provide the background, allowing for the perfect storm that culminates in RA.
Environmental Exposure
As mentioned above, the description of RA has only been recorded since the industrial revolution, supporting a role for the environment. This is further supported by the observation that monozygotic twins are concordant for RA in only about 15% of cases. Smoking was long known to be associated with RA and the presence of rheumatoid factor.10 A number of studies have shown that smoking is strongly associated with ACPA-positive RA.11 The mechanism by which smoking may interact with the SE to produce disease was recently clarified by the observation that smoking was associated with the presence of citrullinated proteins inside alveolar macrophages obtained by lavage of normal smokers.11 Further, the bronchus-associated lymphoid tissue identified in some patients with pulmonary complications of RA produced ACPAs, suggesting that the lung might be the original site for producing ACPAs in some patients.12 Other studies demonstrate that periodontal disease results in the presence of citrullinated proteins locally. These observations identify mechanisms by which environmental exposure may produce citrullinated proteins.
Another environmental element that may contribute to the pathogenesis of RA is the gut microbiome. Studies demonstrate that a variety of forms of experimental arthritis do not develop if the mice are raised in a germ-free environment in which they lack microbial colonization of the intestine. Further, colonization with even a single strain of intestinal bacteria promotes disease expression. A recent article in The Rheumatologist by Drs. Scher and Abramson eloquently reviewed this topic, describing how imbalance of the intestinal microbiota results in an increase of disease-associated lymphocytes including TH1, TH2, and TH17 cells and a reduction of suppressive T regulatory cells, which may promote disease.13 These observations from experimental animals suggests that the intestinal microbiome has the potential to set the immunologic rheostat, perhaps in concert with the SE and other disease-associated SNPs, to promote the induction of the immunologic response that ultimately results in RA.
Preclinical Immune Response
A number of studies demonstrate that rheumatoid factor and ACPAs may be present long before the onset of RA, while a recent study demonstrates that epitope spreading occurs for years prior to the onset of disease and that citrullinated epitopes may be present on multiple autoantigens.5,14 The T cell–dependent induction of ACPAs is mediated by the presentation of citrullinated epitopes bound to cell-surface SEs to antigen-specific T cells, which then provide help to B cells to promote the production of the antibodies.2 Since the autoantibodies may be detected long before the onset of the disease, these observations support the role of the innate immune system as antigen-presenting cells, and T and B lymphocytes in the production of autoantibodies prior to the onset of the disease. Further supporting the role of the innate immune system, IL-6 and soluble TNF receptor II, a proxy for TNF-α, were elevated in plasma samples taken from patients up to 12 years prior to the onset of RA.15 Together, these observations demonstrate that both the innate and adaptive immune systems are active even years prior to the onset of RA.
Role of ACPAs and Immune Complexes in the Development of Clinical Disease
Data have accumulated recently that identify how ACPAs are capable of promoting the development of RA. Circulating immune complexes containing citrullinated fibrinogen have been detected in patients with RA, documenting the ability of citrullinated proteins to form immune complexes.16 Immune complexes formed between citrullinated fibrinogen and ACPAs from patients with RA are capable of activating macrophages to release TNF-α that was mediated not only through Fc receptors but also through TLR4, suggesting that complexes containing citrullinated proteins may be particularly proinflammatory.17 Additionally, these immune complexes containing citrullinated fibrinogen also activated macrophages isolated from the joints of patients with RA.18
It is unclear how circulating antibodies or immune complexes might gain entrance to the joint to initiate clinical disease. A potential answer may come from studies examining the serum-transfer model of arthritis in which serum from K/BxN mice, which develop spontaneous arthritis due to antibodies to a ubiquitous protein called glucose-6-phosphage isomerase (GPI), develop arthritis that begins shortly following serum transfer. Studies have shown that immune complexes formed by the anti-GPI antibodies induce vascular permeability mediated by the release of histamine that permits the pathogenic antibodies and immune complexes to enter the joint.19 Supporting a similar role in RA, immunoglobulin E ACPAs capable of degranulating mast cells, releasing histamine and other proinflammatory mediators, have been identified in RA, suggesting that a similar scenario might occur in early RA, in the transition from preclinical to clinical disease.20
An additional possibility is that the immune complexes are formed within the joint. As mentioned earlier, citrullinated proteins are present in the joints of patients with RA. Further, antibodies to citrullinated proteins as well as ACPA-producing B cells are enriched in the joints of patients with RA, thus facilitating the formation of immune complexes locally.21 As mentioned above, ACPAs containing immune complexes may promote inflammation by activating macrophages. Further, ACPAs from patients with RA are capable of activating both the classical and alternative pathways of complement. Supporting the relevance of this possibility in RA, earlier studies document the local activation of complement within the RA joint, including the presence of increased concentrations of C5a, which is capable of promoting inflammation by recruiting neutrophils and enhancing vascular permeability. Thus, ACPAs containing immune complexes are capable of promoting inflammation by multiple mechanisms, including activation though Fc receptors, TLR receptors, and complement activation.
Rationale for Suspecting a Role for TLRs in RA
Since the production of autoantibodies is T-cell dependent, this implies a role for antigen-presenting cells. Dendritic cell maturation, which enables optimal antigen presentation, is mediated through the activation of TLRs, suggesting a role for TLRs in the initiation of the disease. Now I would like to address how TLR signaling contributes to the persistence and progression of RA. I propose that the initial inflammatory response results in the increased expression and release of molecules that are normally resident within the cell and/or degradation of structural joint components exposing proinflammatory epitopes that may serve as endogenous TLR ligands. Clinical observations support the potential for the engagement of another pathway of joint inflammation once joint damage is initiated.
Earlier studies demonstrate that inflammation may be present in a given joint even in the absence of symptoms or signs of inflammation. Also, initiation of therapy early in RA is more effective than treating longstanding disease. The greater the burden of disease as evidenced by radiographic joint damage at the onset of therapy, the more likely the disease will be progressive and resistant to therapy. These observations suggest that an additional mechanism may become engaged once joint damage has occurred, which is capable of stoking the flames of inflammation, resulting in even greater disease burden. In addition to a potential role in the initiation of the disease, TLRs may contribute to disease progression.
TLRs in the RA Joint
For TLRs to promote inflammation in RA, not only must they be present and functional at the site of inflammation, but TLR ligands must be available locally that are capable of activating through the TLR signaling pathway. Although synovial fibroblasts express TLRs and respond to TLR ligands, cells of the innate immune system, such as macrophages and dendritic cells, are more important for their TLR-mediated responses. We demonstrated that TLR2 and TLR4 on macrophages isolated from the joints of patients with RA are increased compared with their expression on control peripheral blood monocytes or control in vitro differentiated macrophages or macrophages isolated from the joints of patients with other forms of inflammatory arthritis, such as psoriatic arthritis and ankylosing spondylitis.22 In addition, RA synovial fluid macrophages demonstrate a significantly increased response to the TLR2 microbial ligand peptidoglycan and the TLR4 microbial ligand LPS compared to control peripheral blood monocytes or monocyte in vitro differentiated macrophages and macrophages from the joints of patients with psoriatic arthritis and ankylosing spondylitis. This increased sensitivity to signaling though the TLR pathway, which may be mediated through decreased production of the antiinflammatory cytokine IL-10 or increased production of the proinflammatory cytokine interferon γ, may contribute to the progressive inflammation and joint destruction that may be seen in RA, and it may prime patients for progressive inflammation and destruction by RA. It is interesting to speculate that disease-associated TNFAIP3 SNPs might also contribute to this increased TLR-mediated activation in some patients.
Endogenous TLR Ligands in the RA Joint
What are the potential TLR ligands that might drive the chronic inflammation and joint destruction observed in patients with RA? For TLR2 and TLR4 ligands to be active, they must gain access to the extracellular space within the joint, which may be sampled in part by the analysis of synovial fluid. A variety of putative TLR ligands have been described based on their ability to activate through the TLR signaling pathway, including molecules such as HSP-60, -70, and -22, EDA fibronectin, and the structural molecule biglycan. However, we were unable to identify HSP-60 and -70 or biglycan extracellularly in RA synovial fluid and very low endotoxin HSP-60, -70 and -22 failed to activate through TLR2 or 4.23 Further, endotoxin is a contaminant that is capable of activating through TLR4 when recombinant proteins are expressed in Escherichia coli. Therefore, any study that employs recombinant proteins to identify their ability to serve as TLR ligands must take this concern into account.
One way to address this concern is to identify potential endogenous TLR ligands by their ability to directly bind TLR2 or TLR4. We employed the extracellular domain of TLR2 and TLR4 to pull down potential ligands from macrophage cell lysates. With this technique, we identified an endoplasmic reticulum expressed chaperone protein, gp96.23 Other studies identified gp96 as a master chaperone responsible for targeting TLRs to either the endosome or the cell surface.24 In the absence of gp96 in macrophages, the TLRs are expressed but do not localize to their respective locations and are not functional. We observed that gp96 binds to TLR2 and that it is highly expressed in RA synovial tissue and, importantly, extracellularly in RA synovial fluid. The gp96 in the RA synovial fluid is significantly increased compared to fluids from patients with psoriatic arthritis, ankylosing spondylitis, or osteoarthritis.
Using a highly purified, ultralow endotoxin gp96, we were able to demonstrate that gp96 was a potent activator of macrophages, inducing inflammatory cytokines. Additionally, the induction of inflammatory cytokines from RA synovial fluid macrophages by gp96 was significantly greater than observed with peripheral blood monocytes. Further, the concentration of gp96 in the RA synovial fluid was strongly correlated with the expression of TLR2 on RA synovial fluid macrophages, suggesting a self-perpetuating process. Supporting a role for endogenous TLR2 ligands in the progression and joint destruction that may be observed in RA, TLR2 is highly expressed in the leading edge of the RA pannus as it erodes into bone, suggesting that constituents of the bone may contribute to the inflammatory process.25 Consistent with the role of TLR2 in the pathogenesis of RA, incubation of RA synovial tissue explants with a neutralizing antibody to TLR2, suppresses the constitutive expression of TNF-α, suggesting a role for endogenous TLR2 ligands in the ongoing inflammation.26 Further supporting a role for gp96 in RA, our data demonstrate that RA synovial fluid gp96 is capable of activating macrophages mediated through TLR2, and that RA synovial fluid macrophages express significantly increased levels of cell surface gp96, which is capable of activating adjacent macrophages, presumably through TLR2.27 These observations suggest a self-perpetuating cycle, where inflammation results in the increased expression and release of gp96, which then results in further TLR-mediated macrophage activation.
In an attempt to identify other endogenous TLR2 ligands in the synovial tissue of patients with RA, we went fishing, employing a yeast two-hybrid system in which the extracellular domain of TLR2 was the bait, and proteins expressed by a cDNA library constructed from the synovial tissue of patients with longstanding and erosive, active RA were used as the prey. The molecule that most frequently interacted with TLR2 was Snapin, a small molecular-weight protein that is critical for neurotransmission and the transport of autophagosomes within neurons.28 After confirming that Snapin bound to TLR2, we demonstrated that Snapin was capable of activating through TLR2. Snapin is highly expressed in RA synovial tissue examined by immunohistochemistry and qRT-PCR.29 There was a significant correlation between Snapin in the RA synovial tissue by qRT-PCR and clinical disease activity determined by number of swollen joints. Supporting the relevance of Snapin to the pathogenesis of RA, a neutralizing antibody to Snapin suppressed the ability of some RA synovial fluids to activate macrophages. These observations identify gp96 and Snapin as endogenous TLR2 ligands that contribute to the pathogenesis of RA.
There are convincing data that endogenous TLR4 ligands may also contribute to the pathogenesis of RA. RA synovial fluids are capable of activating human embryonic kidney cells that are engineered to specifically respond to TLR4 ligands.30 Additionally, the constitutive expression of inflammatory cytokines by RA synovial tissue explant cultures was suppressed by a TLR4 inhibitor. These observations suggest the presence of endogenous TLR4 ligands, capable of driving inflammation in the synovial tissue and fluid of patients with RA. Although studies employing fluids or tissues from patients with RA have not functionally identified a specific pathogenic endogenous TLR4 ligand, tenascin C has emerged as an interesting candidate.
Tenacin C is an extracellular matrix glycoprotein that is not generally expressed in adult tissue but is highly expressed in the rheumatoid joint. Tenacin C, and specifically the region of the molecule called the fibrinogen-like bulb, mediates macrophage activation and induces proinflammatory cytokine production when incubated in RA synovial tissue explant cultures.31 Interestingly, mice deficient in tenascin C, following the induction of immune complex–mediated arthritis, exhibit a more rapid resolution of the arthritis compared with mice that expressed tenascin C, suggesting that the presence of tenascin C promotes ongoing inflammation.
Relevance and Potential Therapeutic Strategy
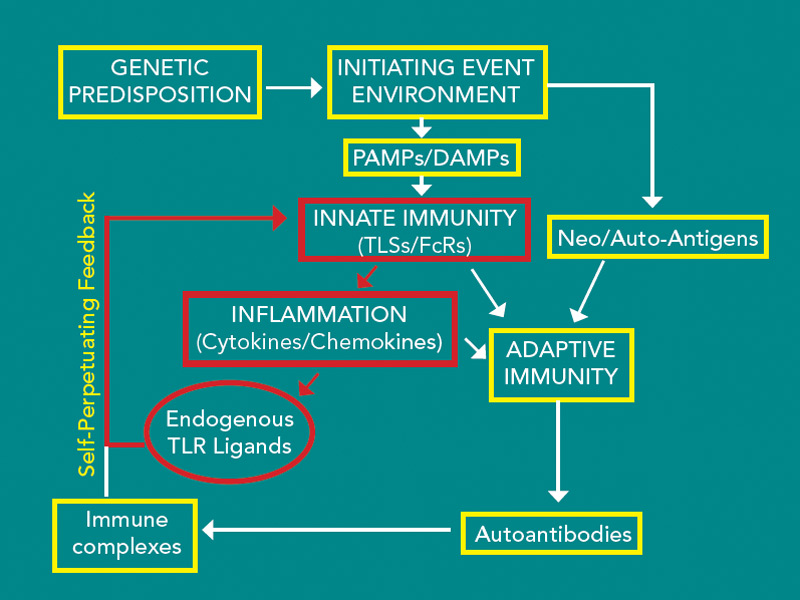
Together, these observations suggest that environmental exposure results in the generation of citrullinated neo-antigens and in the genetically susceptible individual possessing a SE, preclinical disease-associated antibodies are generated. As the repertoire expands, these antibodies appear to result in the activation of innate immunity identified by the production of proinflammatory cytokines and chemokines. By mechanisms yet to be defined, this response becomes apparent in the joints, and clinical disease begins. The induced inflammatory response appears capable of inducing endogenous TLR ligands such as gp96, Snapin, and tenascin C, which may be capable of promoting progressive inflammation, potentially leading to joint destruction. The clinical observation that treating early disease is more effective than treating disease once structural joint damage has occurred may be related to the induction of endogenous TLR ligands, which results in a self-perpetuating process (see Figure 2). At what point in the clinical course endogenous TLR ligands might contribute to the process is not clear. Studies of the synovial tissue of patients with early RA generally cannot be distinguished from those with long standing disease. However, the presence of potential endogenous TLR ligands has not been examined early in the disease.
Recent studies suggest a number of potentially important endogenous TLR ligands. Therefore, neutralization of individual molecules does not appear to be a realistic approach. Strategies that target TLR2 or TLR4, employing neutralizing monoclonal antibodies or even recombinant TLR molecules, fashioned after etanercept, might be possible. In fact, the extracellular domain of TLR2 was effective at suppressing TLR2-mediated inflammation.32 Additional methods to target these pathways include the use of inhibitors that bind the extracellular domain of TLR2 or TLR4, such as has been used in explant cultures.26,30 Additionally, small molecule inhibitors of the Myd88/IRAK pathway might selectively block TLR signaling, leaving other pathways intact. Supporting the value of targeting the TLR signaling pathways, a preliminary study employing a small molecular-weight heat shock protein called chaperonin 10, an inhibitor of TLR signaling, was effective and safe in a small group of patients with RA.33 Together, these observations suggest that there is a Toll that is paid in the persistent and progressive inflammation and joint destruction observed in RA. the rheumatologist
Dr. Pope is chief of the division of medicine–rheumatology at Northwestern University Feinberg School of Medicine in Chicago.
References
- Hansson GK, Edfeldt K. Toll to be paid at the gateway to the vessel wall. Arterioscler Thromb Vasc Biol. 2005;25:1085-1087.
- Snir O, Rieck M, Gebe JA, et al. Identification and functional characterization of T cells reactive to citrullinated vimentin in HLA-DRB1*0401-positive humanized mice and rheumatoid arthritis patients. Arthritis Rheum. 2011;63:2873-2783.
- Stastny P. Mixed lymphocyte cultures in rheumatoid arthritis. J Clin Invest.1976;57:1148-1157.
- Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205-1213.
- Rantapaa-Dahlqvist S, de Jong BA, Berglin E, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48:2741-2749.
- Raychaudhuri S, Sandor C, Stahl EA, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet. 2012;44:291-296.
- Zhang J, Zahir N, Jiang Q, et al. The autoimmune disease-associated PTPN22 variant promotes calpain-mediated Lyp/Pep degradation associated with lymphocyte and dendritic cell hyperresponsiveness. Nat Genet. 2011;43:902-907.
- Menard L, Saadoun D, Isnardi I, et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest. 2011;121:3635-3644.
- Matmati M, Jacques P, Maelfait J, et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat Genet. 2011;43:908-912.
- Silman AJ, MacGregor AJ, Thomson W, et al. Twin concordance rates for rheumatoid arthritis: results from a nationwide study. Br J Rheumatol. 1993;32:903-907.
- Klareskog L, Stolt P, Lundberg K, et al. A new model for an etiology of rheumatoid arthritis: Smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2006;54:38-46.
- Rangel-Moreno J, Hartson L, Navarro C, Gaxiola M, Selman M, Randall TD. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J Clin Invest. 2006;116:3183-3194.
- Scher JU, Abramson SB. The microbiome. The Rheumatologist. 2011;5(11):1, 32-35.
- van de Stadt LA, de Koning MH, van de Stadt RJ, et al. Development of the anti-citrullinated protein antibody repertoire prior to the onset of rheumatoid arthritis. Arthritis Rheum. 2011;63:3226-3233.
- Karlson EW, Chibnik LB, Tworoger SS, et al. Biomarkers of inflammation and development of rheumatoid arthritis in women from two prospective cohort studies. Arthritis Rheum. 2009;60:641-652.
- Zhao X, Okeke NL, Sharpe O, et al. Circulating immune complexes contain citrullinated fibrinogen in rheumatoid arthritis. Arthritis Res Ther. 2008;10:R94.
- Sokolove J, Zhao X, Chandra PE, Robinson WH. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcgamma receptor. Arthritis Rheum. 2011;63:53-62.
- Laurent L, Clavel C, Lemaire O, et al. Fcgamma receptor profile of monocytes and macrophages from rheumatoid arthritis patients and their response to immune complexes formed with autoantibodies to citrullinated proteins. Ann Rheum Dis. 2011;70:1052-1059.
- Binstadt BA, Patel PR, Alencar H, et al. Particularities of the vasculature can promote the organ specificity of autoimmune attack. Nat Immunol. 2006;7:284-792.
- Schuerwegh AJ, Ioan-Facsinay A, Dorjee AL, et al. Evidence for a functional role of IgE anticitrullinated protein antibodies in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2010;107:2586-2591.
- Snir O, Widhe M, Hermansson M, et al. Antibodies to several citrullinated antigens are enriched in the joints of rheumatoid arthritis patients. Arthritis Rheum. 2010;62:44-52.
- Huang Q, Ma Y, Adebayo A, Pope RM. Increased macrophage activation mediated through toll-like receptors in rheumatoid arthritis. Arthritis Rheum. 2007;56:2192-2201.
- Huang QQ, Sobkoviak R, Jockheck-Clark AR, et al. Heat shock protein 96 is elevated in rheumatoid arthritis and activates macrophages primarily via TLR2 signaling. J Immunol. 2009;182:4965-4973.
- Yang Y, Liu B, Dai J, et al. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity. 2007; 26:215-226.
- Seibl R, Birchler T, Loeliger S, et al. Expression and regulation of Toll-like receptor 2 in rheumatoid arthritis synovium. Am J Pathol. 2003;162:1221-1227.
- Ultaigh SN, Saber TP, McCormick J, et al. Blockade of Toll-like receptor 2 prevents spontaneous cytokine release from rheumatoid arthritis ex vivo synovial explant cultures. Arthritis Res Ther. 2011;13:R33.
- Huang QQ, Koessler RE, Birkett R, et al. Gp96 perpetuates the persistent inflammation of rheumatoid arthritis. Arthritis Rheum. 2012 Jul 6. [Epub ahead of print]
- Yuzaki M. Snapin snaps into the dynein complex for late endosome-lysosome trafficking and autophagy. Neuron. 2010;68:4-6.
- Shi B, Huang Q, Tak PP, et al. SNAPIN: An endogenous toll-like receptor ligand in rheumatoid arthritis. Ann Rheum Dis. 2012 Apr 20. [Epub ahead of print]
- Abdollahi-Roodsaz S, Joosten LA, Koenders MI, et al. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest. 2008;118:205-216.
- Midwood K, Sacre S, Piccinini AM, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. 2009;15:774-780.
- Raby AC, Le Bouder E, Colmont C, et al. Soluble TLR2 reduces inflammation without compromising bacterial clearance by disrupting TLR2 triggering. J Immunol. 2009; 183:506-517.
- Vanags D, Williams B, Johnson B, et al. Therapeutic efficacy and safety of chaperonin 10 in patients with rheumatoid arthritis: A double-blind randomised trial. Lancet. 2006;368:855-863.