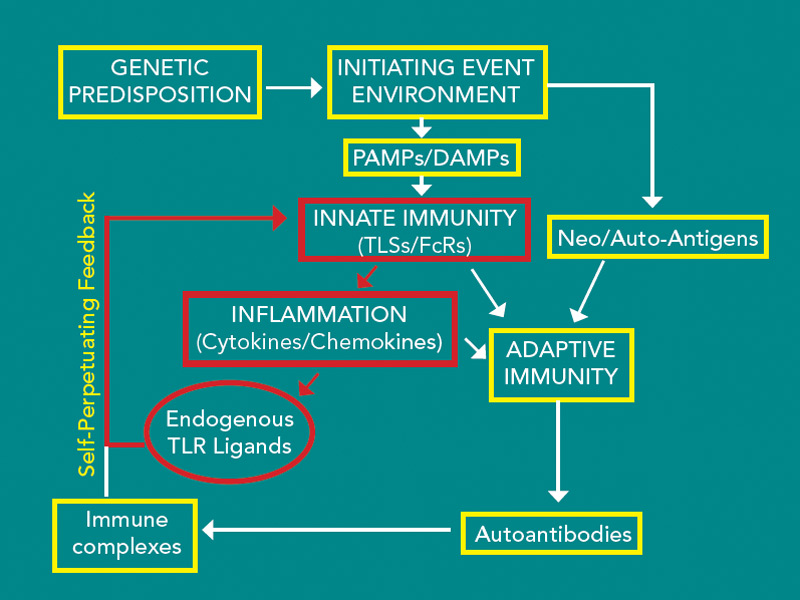
Recent studies suggest a number of potentially important endogenous TLR ligands. Therefore, neutralization of individual molecules does not appear to be a realistic approach. Strategies that target TLR2 or TLR4, employing neutralizing monoclonal antibodies or even recombinant TLR molecules, fashioned after etanercept, might be possible. In fact, the extracellular domain of TLR2 was effective at suppressing TLR2-mediated inflammation.32 Additional methods to target these pathways include the use of inhibitors that bind the extracellular domain of TLR2 or TLR4, such as has been used in explant cultures.26,30 Additionally, small molecule inhibitors of the Myd88/IRAK pathway might selectively block TLR signaling, leaving other pathways intact. Supporting the value of targeting the TLR signaling pathways, a preliminary study employing a small molecular-weight heat shock protein called chaperonin 10, an inhibitor of TLR signaling, was effective and safe in a small group of patients with RA.33 Together, these observations suggest that there is a Toll that is paid in the persistent and progressive inflammation and joint destruction observed in RA. the rheumatologist