The Critical Need to Control Complement Activation
Because of the design of the feedback loop coupled with its effector capabilities, tight regulation is critical. To this end, nearly half of the proteins of the complement system act to mitigate its effects in plasma (fluid phase, no target) and on healthy host cells (wrong target).14 However, once cells are injured, limited complement activation is desirable to facilitate tissue repair and to provide for safe removal of debris—the so-called “targeted and restricted complement activation.”15
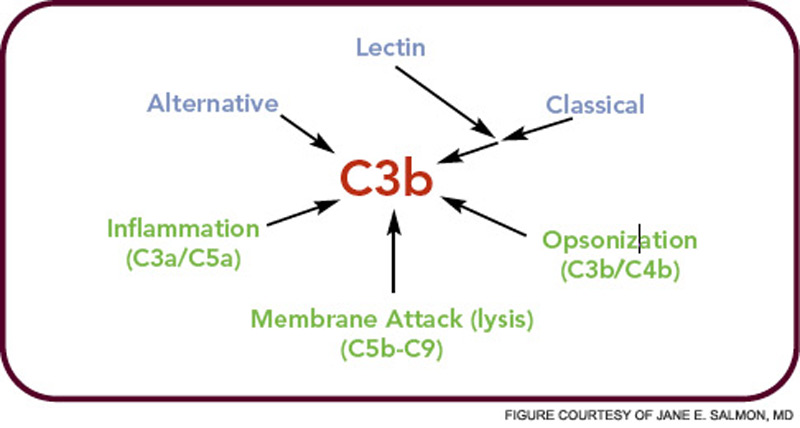
The critical step of C3 cleavage is controlled by regulators that operate through two processes. One mechanism is cofactor activity in which a protein such as the plasma protein factor H (FH) or the nearly ubiquitously expressed membrane cofactor protein (MCP; CD46) binds to C3b (see Figure 3, p. 17). This binding allows a third player, a serine protease of plasma known as factor I, to then degrade C3b. The second process is called decay accelerating activity, in which the catalytic (serine protease) domain of the multiunit C3 convertases is disassociated. These protective mechanisms in large part explain how the complement system separates self from nonself; that is, healthy human tissue is protected, and homeostasis maintained by a constitutively expressed array of regulators on cells and by proteins circulating in plasma. Microbes and damaged cells are not so well shielded. However, a common theme is for opportunistic organisms, especially those that invade the bloodstream, to produce virulence factors that bind the host’s own plasma inhibitors or to synthesize their own CIP mimics.16
Dysfunctions of Regulation Leading to Disease Associations
Cellular wastes, including apoptotic and necrotic cells, need to be efficiently eliminated.17 A simple etiologic explanation for systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) focuses on the consequences of mishandling of DNA/ RNA protein complexes in the former and citrullinated proteins in the latter. Such biological “garbage” can become an immunogen for an adaptive immune response or a nidus for chronic inflammation. Deposition of debris is associated with several of the most common, serious, and lethal illnesses in the developed world: heart attacks and strokes secondary to atherosclerosis (oxidized lipids); dementia in Alzheimer’s disease from misfolded proteins (amyloid); and vision loss in AMD secondary to lipofuscin pigments (drusen). Chronic tophaceous gout would also be in epidemic form, if it were not for our ability to control uric acid levels.