Upon arrival, she was febrile to 102.2ºF, with a heart rate of 100 beats per minute, blood pressure of 90–100/40–50, and a respiratory rate of 24 with normal oxygen saturation on room air. She was pale in appearance. Her abdomen was soft with tenderness in the left upper quadrant and epigastrium, and the spleen was palpable. Her musculoskeletal exam revealed tenderness at the metacarpophalangeal and proximal interphalangeal joints of both hands, without swelling.
Laboratory studies included a white blood cell count of 2.4 K/µL, hemoglobin of 6.7 g/dL and platelet count of 84 K/µL. Her CRP was elevated to 90 mg/L and her ESR was 33 mm/h. Her ferritin was 543 ng/mL and triglyceride was
325 mg/dL. The remainder of her labs were unremarkable. She had a negative serological workup for ANA, ANCA, RF, anti-CCP and antibodies to RNP, Jo-1, Ro and La. SPEP revealed no abnormalities.
A chest X-ray demonstrated small opacities at the base of the left lung consistent with atelectasis, as well as a small left pleural effusion. A CT of the chest revealed no evidence of intrathoracic infection or malignancy, but trace bilateral pleural effusions and pericardial effusions were noted. A CT of the abdomen/pelvis with contrast demonstrated moderate splenomegaly (16.4 cm), without evidence of infection or malignancy.
In summary, the patient was a 48-year-old woman with three months of daily fevers to 103ºF, night sweats and intermittent abdominal discomfort, now with pancytopenia, moderate splenomegaly, small pericardial and pleural effusions with negative infectious disease workup and normal bone marrow biopsy.
Diagnosis
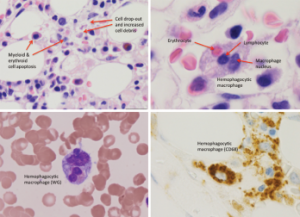
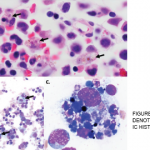
Figure 1: Bone Marrow Biopsy
Hemophagocytosis, with cell apoptosis, cell drop-out and increased cell debris, is shown.
When we met our patient, systemic rheumatic diseases—such as rheumatoid arthritis, systemic lupus erythematosus, systemic sclerosis and mixed connective tissue disease—were considered unlikely given the lack of typical clinical features and negative autoantibody profile. Malignancy also seemed improbable given her negative workup and lack of lymphadenopathy.
Although her CBC showed pancytopenia, the peripheral blood smear was unremarkable. After further discussion with our hematology consultants, a repeat bone marrow biopsy was performed. It demonstrated hypercellularity with trilineage maturing hematopoiesis, stromal damage, cell dropout and increased macrophage phagocytic activity (see Figure 1), which raised the possibility of hemophagocytic lymphohistiocytosis (HLH).
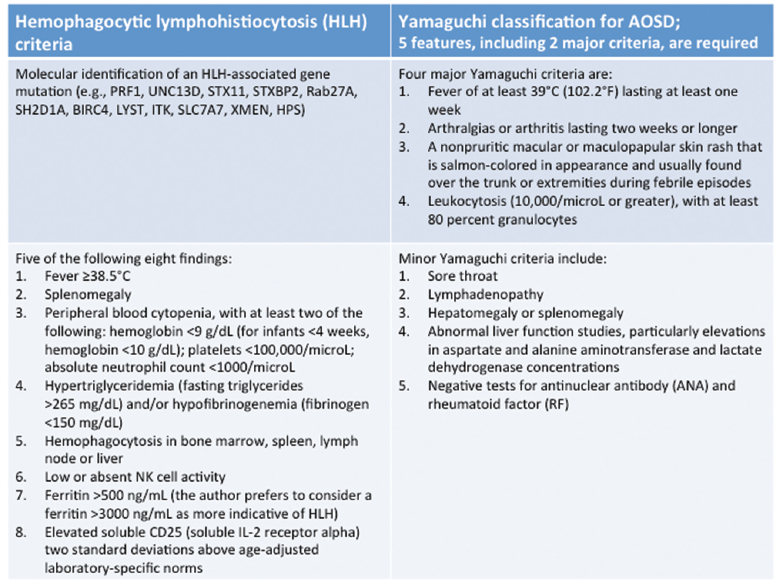
Reviewing the criteria for HLH, we noted that she met five out of the eight criteria, including fever, splenomegaly, pancytopenia, hypertriglyceridemia, a ferritin greater than 500, as well as hemophagocytosis in the bone marrow (see Table 1).2-4
The suspicion of HLH led to the question of etiology and whether it might be related to adult-onset Still’s disease (AOSD). She fulfilled the Yamaguchi criteria for AOSD, including two major criteria (fever and arthralgias lasting two or more weeks) as well as three minor criteria (splenomegaly, abnormal liver function studies with elevations and negative tests for ANA and RF) (see Table 1).2-4
Click for larger image
Table 1: HLH (left) & AOSD (right) Criteria
The patient remained febrile to 104ºF with abdominal pain, which had now progressed to involve her left lower quadrant and right upper quadrant with diffuse myalgias, fatigue and diffuse arthralgias in her hands. Given her ongoing symptoms, suspicion of HLH due to AOSD and otherwise negative evaluation, she was administered pulse doses of steroids 1 g IV solumedrol for three days followed by daily prednisone (1 mg/kg).
She continued to have worsening fatigue, myalgias and arthralgias involving the elbows and wrists, as well as left upper quadrant and left lower quadrant abdominal pain with fevers to 101.8ºF. Her labs were significant for worsening pancytopenia with leukopenia to 2.8 K/µL, thrombocytopenia to 58 K/µL and anemia with hemoglobin 5 g/dL. Additionally, it was noted that her ferritin increased to 1,197 ng/mL and her triglyceride increased to 560 mg/dL. She received three additional days of high-dose steroids with methylprednisolone 1,000 mg IV/day, and anakinra by subcutaneous injection (100 mg daily) was initiated. She again experienced worsening fatigue and night sweats when her steroids were transitioned to oral prednisone, so the dose of anakinra was increased to 200 mg daily.
Due to her persistent symptoms, there was concern that the driving force behind her HLH may not be AOSD as originally suspected. While this was being deliberated, her soluble interleukin (IL) 2 receptor alpha (sCD25 or sIL-2R) level was reported to be elevated to 9959. Past studies by Tsuji et al showed that the mean serum ratio of sIL2R:ferritin of patients with HLH secondary to lymphoma was found to be greater than the ratio in benign disease-associated HLH. Our patient’s serum ratio of serum soluble interleukin 2 receptor (sIL-2R):ferritin was markedly elevated at 8.3; therefore, malignancy-associated HLH was reconsidered despite her prior negative evaluation.
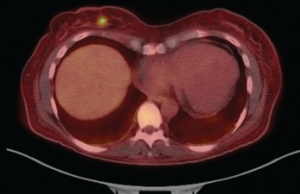
Figure 2: Fused PET-CT Image
The fused PET-CT shows the FDG-avid, right breast lesion measuring 7×4 mm, which is concerning for malignancy.
For further workup, she had a PET scan performed, which revealed an FDG-avid, 7 mm right breast lesion (see Figure 2). A mammogram showed an echogenic mass, concerning for malignancy. An ultrasound-guided core biopsy of this lesion revealed a diffuse large B cell lymphoma (see Figure 3), with the breast as the likely primary site (high-grade breast lymphoma). The neoplastic cells were found to be diffusely immunoreactive for CD20, co-expressed BCL6, BCL2 and MUM1, and were negative for CD10, CD43 and BCL1.
Soon after this diagnosis was established, she completed six cycles of intrathecal methotrexate with corticosteroids, EPOCH (etoposide, prednisolone, oncovin or vincristine, cyclophosphamide, hydroxydaunorubicin) and rituximab. Her symptoms, as well as her pancytopenia, resolved. Repeat PET scans over the past year have demonstrated no evidence of disease.
Discussion
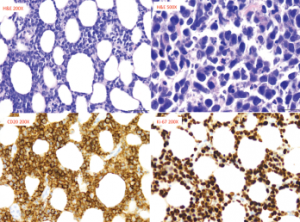
Figure 3: Pathology from Right Breast Core Biopsy
The biopsy revealed diffuse large B cell lymphoma, with neoplastic cells, which are diffusely immunoreactive for CD20. These cells also co-expressed BCL6, BCL2 and MUM1, but were negative for CD10, CD43 and BCL1.
HLH is a syndrome of excessive immune activation of mononuclear phagocytes with elevated cytokines, which can result in a difficult to control hyperinflammatory response.1 It can present as a febrile illness associated with multiple organ involvement. Some common presentations include fever of unknown origin, hepatitis, encephalitis or a mimic of common infections. HLH can be diagnosed by the molecular identification of an HLH-associated gene mutation or the presence of five out of eight criteria, as listed in Table 1.2-4 Macrophage activation syndrome (MAS) is HLH that usually occurs in association with a rheumatologic condition, such as AOSD.
Based on the HLH-2004 trial, there are two types of HLH: 1) primary (or familial) HLH, which occurs due to a genetic mutation and 2) secondary (or acquired) HLH, which is usually associated with an infection, malignancy or autoimmune disorder. HLH is usually triggered by a variety of events that disrupt the homeostasis of the immune system. The pathognomonic finding in HLH is the presence of hemophagocytosis, which can be found in the bone marrow, lymph nodes, spleen or liver.1 HLH can be life-threatening and catastrophic with an overall mortality of 58–75%.1 Hence, it is important to establish the diagnosis as quickly as possible and initiate immediate treatment. This is often challenging given the rareness of this syndrome, the variable spectrum of clinical presentation, and the lack of specific clinical and laboratory findings to diagnose this condition.
Our patient was initially thought to have AOSD. The Yamaguchi criteria (see Table 1) are used to aid in the diagnosis of AOSD and require the presence of five features, with at least two being major diagnostic criteria.4 Our case met five criteria, but did not have findings of the classic evanescent, salmon-colored rash or leukocytosis. Typically, patients with AOSD have leukocytosis, unless the disease has progressed to MAS/HLH, at which point they are noted to be cytopenic.
HLH & Malignancy
HLH has been associated with lymphoid cancers, including T, NK and anaplastic large-cell lymphomas, as well as leukemias, which include B cell lymphoblastic leukemia and solid tumors.5 The diagnosis of HLH may precede the finding of the malignancy, but this is rare.6 Lymphoma-associated hemophagocytic syndrome (LAHS) is a type of HLH that accounts for approximately 40% of adult-onset secondary HLH and has a poor prognosis.7
A retrospective chart review study evaluated the clinical and laboratory findings, as well as the prognostic factors, of adult HLH in a cohort of 74 patients from 2003 to 2014; 73 patients, with a median age of 51 years, met the HLH-2004 diagnostic criteria. Fever, cytopenias and elevated ferritin were noted in greater than 85% of cases. Infection was the associated etiology of HLH in 41%; whereas, 28.8% of cases were due to malignancy. To a lesser extent, there was an association of HLH with autoimmune conditions (7%), post-solid organ transplantation (3%) and primary immunodeficiency (1.4%); the remaining cases were labeled idiopathic (18%). This study demonstrated that cases with malignancy-associated HLH had a significantly worse survival compared with patients with non-malignancy-associated HLH (median overall survival 1.1 vs. 46.5 months, respectively; P<0.0001). Analysis of these patients’ 30-day mortality rates showed that a ferritin greater than 50,000 µg/L was the only poor predictor.1
In another retrospective study including 162 patients diagnosed with HLH, 56% of cases were associated with a hematologic malignancy, especially non-Hodgkin’s lymphomas; only 70% of patients had features of hemophagocytosis in the bone marrow.8
An elevated sIL-2R two standard deviations above age-adjusted laboratory-specific norms is included in the diagnostic criteria for HLH. This is a marker for the presence of activated lymphocytes, which is usually measured by enzyme-linked immunosorbent assay.1 A normal level of sIL-2R for adults depends on the number of cells producing the IL-2 receptor and the number of molecules per cell. Although there is wide variation in the normal range of sIL-2R between laboratories, a value greater than two standard deviations above the normal range for a particular laboratory (or 2,400 U/mL in pediatric data sets9) has been found to be diagnostically useful.
A retrospective analysis of the laboratory findings of 21 patients with HLH (11 lymphoma-associated HLH and 10 benign disease-associated HLH) found the mean sIL-2R levels were significantly higher in the lymphoma-associated group (13,451 U/mL vs. 4,176 U/mL, P=0.0031), and ferritin levels were higher in the benign disease-associated HLH group (20,462 ng/mL vs. 2,561 ng/mL, P=0.0031).7 The mean serum sIL-2R:ferritin ratio of patients with lymphoma-associated disease was markedly higher than that of patients with benign disease-associated HLH (8.5 vs. 0.7, P=0.0004).7
This study suggests that the sIL‑2R:ferritin ratio can be useful as a marker in the diagnosis of LAHS.7
Treatment of HLH
The aim of therapy in patients with HLH is to suppress inflammation by destroying the responsible immune cells. Based on the HLH-94 protocol, treatment for acutely ill patients with primary HLH should begin with eight weeks of induction therapy with etoposide and dexamethasone, with intrathecal methotrexate therapy for any central nervous system involvement.
It is important to start therapy as soon as the diagnosis is recognized given the high mortality of untreated disease. Once induction is completed, patients are weaned off therapy while those patients who are not improving are continued on therapy until they can be offered allogeneic hematopoietic cell transplantation (HCT). Generally, stem cell transplant is needed for those patients with an HLH mutation, central nervous system disease or disease relapse.10 Median survival has been shown to be 54% at 6.2 years with treatment based on the HLH-94 protocol.10
In cases of secondary HLH, it’s essential to treat the underlying triggering condition, such as the infection, rheumatic disease or malignancy. Response to therapy is followed by monitoring the disease specific markers.
Conclusion
HLH is a life-threatening condition that requires prompt diagnosis and management. In patients with suspected HLH, more than one bone marrow biopsy may be needed for diagnosis, as in our patient. Patients with HLH should undergo thorough screening for malignancy, starting with age- and gender-
appropriate testing and imaging tests. When there is a strong suspicion for malignancy, it may be appropriate to consider PET scanning. A marked elevation in the ratio of sIL-2R to ferritin may suggest the presence of lymphoma-associated HLH rather than HLH related to benign disease.
 Anita Laloo, MB, BS, MPH, is a recent graduate of the Beth Israel Deaconess Medical Center Rheumatology Fellowship in Boston.
Anita Laloo, MB, BS, MPH, is a recent graduate of the Beth Israel Deaconess Medical Center Rheumatology Fellowship in Boston.
German Pihan, MD, is the chief of Diagnostic Hematopathology Service and director of the Hematopathology Fellowship Training Program at Beth Israel Deaconess Medical Center in Boston.
 Robert H. Shmerling, MD, is the clinical chief of Rheumatology and Robinson Firm Chief at Beth Israel Deaconess Medical Center in Boston and a senior editor at Harvard Health Publications.
Robert H. Shmerling, MD, is the clinical chief of Rheumatology and Robinson Firm Chief at Beth Israel Deaconess Medical Center in Boston and a senior editor at Harvard Health Publications.
References
- Otrock ZK, Eby CS. Clinical characteristics, prognostic factors, and outcomes of adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol. 2015 Mar;90(3):220–224.
- Jordan MB, Allen CE, Weitzman S, et al. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011 Oct 13;118(15):4041–4052.
- Henter JI, Horne A, Arico M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007 Feb;48(2):124–131.
- Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992 Mar;19(3):424.
- Miyahara M, Sano M, Shibata K, et al. B-cell lymphoma-associated hemophagocytic syndrome: Clinicopathological characteristics. Ann Hematol. 2000 Jul;79(7):378.
- Chang TY, Jaffray J, Woda B, et al. Hemophagocytic lymphohistiocytosis with MUNC13-4 gene mutation or reduced natural killer cell function prior to onset of childhood leukemia. Pediatr Blood Cancer. 2011 May;56(5):856–858.
- Tsuji T, Hirano T, Yamasaki H, et al. A high sIL-2R/ferritin ratio is a useful marker for the diagnosis of lymphoma-associated hemophagocytic syndrome. Ann Hematol. 2014 May;93(5):821–826.
- Rivière S, Galicier L, Coppo P, et al. Reactive hemophagocytic syndrome in adults: A retrospective analysis of 162 patients. Am J Med. 2014 Nov;127(11):1118.
- Tothova Z, Berliner N. Hemophagocytic syndrome and critical illness: New insights into diagnosis and management. J Intensive Care Med. 2015 Oct;30(7):401–412.
- Trottestam H, Horne A, Aricò M, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011 Oct 27;118(17):4577–4584.