In this case of a neuromyelitis optica spectrum disorder, the patient presented with bilateral vision loss.
Lightspring/shutterstock.com
Case Presentation
The patient was a 42-year-old African American female diagnosed with systemic lupus erythematosus (SLE) based on the findings of polyarthritis, malar and discoid rash, fatigue, positive double-stranded DNA (dsDNA) ribonucleoprotein and Smith antibodies, and low serum complement levels. Her SLE had been well controlled on hydroxychloroquine 400 mg daily, oral methotrexate 25 mg weekly and prednisone 10 mg daily.
She felt well until two days prior to her presentation, when she started experiencing neck pain, which was associated with nausea, vomiting and blurred vision in her left eye, which rapidly progressed to bilateral vision loss. She could visualize only shadows of objects less than one foot from her face. She described one episode of fever to 102º Fahrenheit. She denied any additional symptoms, such as numbness, tingling, weakness, slurred speech, new rash, cough, shortness of breath, abdominal pain, diarrhea, constipation, dysuria or change in urine frequency or color.
The laboratory work-up was notable for a white blood cell count of 3.1×109/L (range 3.2–9.8) with 84% neutrophils, hemoglobin 11.6 g/dL (range 12–15.5), sedimentation rate of 20 mm/hr, C-reactive protein of 1.2 mg/dL (range <0.6), dsDNA 263 IU/mL (range <100, patient’s baseline above 200), C3 of 69 mg/dL (range 86–208, patient’s baseline 75–85), and C4 of 10 mg/dL (range 13–47, patient’s baseline 12–14). Urinalysis was negative for blood or protein.
An MRI of her brain with MRA/MRV (see Figure 1) demonstrated “increased FLAIR signal in the optic chiasm and bilateral intracranial optic nerves and proximal optic radiations, without enhancement. There was possible enlargement of the intracranial optic nerves and optic chiasm.” The MRV of the brain and an MRI of the spine were unremarkable.
During her early hospital course, she remained afebrile and continued on empiric treatment for bacterial meningitis while culture results were monitored. Methotrexate was held. Her headache and nausea completely resolved, but the visual loss persisted. After three days of negative cultures, an infection was felt to be less likely, and she was treated with solumedrol 1,000 mg IV for three days. A repeat lumbar puncture demonstrated an opening pressure of 29 cm CSF, 9 nucleated cells/uL with 84% lymphocytes, glucose of 68 mg/dL and protein 22 mg/dL. Bacterial cultures from blood and CSF remained negative. Viral and fungal studies including enterovirus, cryptococcal, varicella zoster, cytomegalovirus, herpes simplex and others were negative.
Diagnosis & Treatment
Testing for IgG to Aquaporin-4 in both the serum and CSF was positive, leading to a diagnosis of neuromyelitis optica (NMO).
She subsequently received five courses of plasmapheresis followed by rituximab 500 mg IV given once. Antibiotics were discontinued. It was decided to replace methotrexate with mycophenolate mofetil. By discharge, she felt that her vision was improving, albeit very slowly. She could track objects and people in the room, but she was still unable to read any letters on a Snellen chart. She was continued on hydroxychloroquine and discharged on a dose of prednisone 60 mg daily with a prolonged taper.
Discussion
Neuromyelitis optica spectrum disorders (NMOSDs), also known as Devic disease, are autoinflammatory, demyelinating conditions affecting the central nervous system. The pathogenesis is thought to be directly linked to the highly specific Aquaporin-4 antibody (AQP4-IgG), which targets the water channel most common in astrocytes. With the discovery of this antibody, it is now evident that NMOSD is not simply a subtype of multiple sclerosis (MS), but rather a distinct entity. Typical symptoms include blindness or weakness, depending on the location of the demyelinating lesion. The primary cause of mortality is severe ascending cervical myelitis or respiratory failure from brainstem lesions.
This rare disorder is estimated to affect fewer than 1 to 4.4 of 100,000 individuals in Europe and North America.1 It typically affects individuals between the ages of 35 and 45 and more commonly affects women, particularly patients who are seropositive for AQP4-IgG (10:1 female preponderance) vs. seronegative patients (approximately 2:1). There appears to be a distinct presentation with those who are seropositive exhibiting a chronic relapsing course, whereas seronegative patients often experience a monophasic episode.
The CSF analysis of patients with NMOSD is variable. White blood cell counts in CSF are frequently normal or only minimally elevated; however, very high cell counts above 100/uL similar to our patient have been described.2 The presence of neutrophils or eosinophils greater than 5/uL argues in favor of NMOSD over MS. The lack of oligoclonal bands is supportive of the diagnosis because it is seen in less than 20% of patients with NMOSD; however, they can be transiently noted during an attack.
In 2015, a revised international consensus criteria for the diagnosis of NMOSD proposed by the American Academy of Neurology included criteria using the results of testing for AQP4-IgG.2 The diagnostic criteria state the patient should demonstrate at least one other core clinical characteristic (see Table 1) if AQP4-IgG is positive. If AQP4-IgG is negative or unknown, then the patient should have at least two core clinical characteristics, with one of them being among the first three characteristics mentioned in Table 1.

(click for larger image)
TABLE 1: Core Clinical Characteristics for NMOSD
If AQP4-IgG is positive, then the patient must exhibit one of the six clinical characteristics listed. If AQP4-IgG is negative or unknown, two clinical features are required, but one must be a characteristic identified in bold. Adapted from Wingerchuk et al. 2015. Ref. No. 2.
In addition, it is imperative to exclude alternative diagnoses, such as infection, malignancy, MS and sarcoidosis. This is especially important to consider in situations in which the clinical presentation demonstrates a more gradual neurologic course with clinical worsening otherwise not related to attacks; when the CSF analysis demonstrates oligoclonal bands; when there are imaging features more typical of MS. The expected MRI findings of optic neuritis demonstrate increased signal in the optic nerve on T2-weighted images, with fat suppression, with gadolinium enhancement seen on T1.
NMOSD can be related to other autoimmune/rheumatologic conditions, as well. It is estimated that 30–50% of NMOSD patients have laboratory or clinical evidence of these diseases.1 One retrospective study conducted at the Mayo Clinic evaluated 153 U.S. patients with NMOSD and found that there was coexisting autoimmune disease in 62 (40.5%) of the patients—autoimmune thyroid disease (17%), ulcerative colitis (2.6%), SLE (2%), Sjögren’s syndrome (2%), rheumatoid arthritis (1.3%), myasthenia gravis (1.3%) and idiopathic thrombocytopenic purpura (1.3%), among others.3 Because some of these conditions, such as SLE, may cause visual loss, one should carefully evaluate patients to rule out the diagnostic possibilities, especially in those patients who are negative for AQP4-IgG.
NMOSDs, also known as Devic disease, are autoinflammatory, demyelinating conditions affecting the central nervous system. The pathogenesis is thought to be directly linked to the highly specific Aquaporin-4 antibody (AQP4-IgG), which targets the water channel most common in astrocytes.
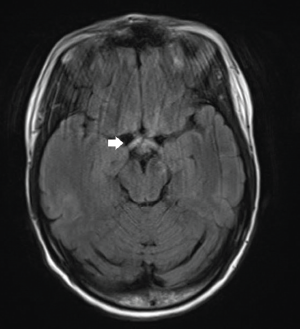
FLAIR sequence demonstrates increased signal in the optic chiasm and proximal optic radiations, as well as optic nerves.
Three studies in the U.S. and Europe have found that the frequency of AQP4-IgG did not differ dramatically between cases of NMOSD alone and NMOSD associated with another rheumatologic condition; both populations exhibited high proportions of seropositivity.4 In the absence of symptoms of NMOSD, Aquaporin-4 antibody was typically absent; however, a separate study in Greece looking at 89 patients with SLE without any neurologic disease found two patients who were seropositive for AQP4-IgG in multiple serum samples dating back as far as 11 years prior to study entry.5 These patients were also reevaluated for neurologic disease and subsequently obtained neuroimaging with MRI that still did not show any evidence of typical NMOSD lesions.
The primary goals of treatment of NMOSD are to recover any lost neurologic function and to prevent relapses. Most studies evaluating treatment recommendations are limited by their small sample size, and few are prospective in nature. Initial treatment to recover neurologic function is typically anchored by high-dose intravenous steroid.1 If there is an insufficient response, then plasma exchange is pursued next. If symptoms still do not demonstrate improvement, intravenous immunoglobulins have been administered.
Long-term immunosuppression is used to prevent relapses. Multiple options exist, but rituximab is a commonly used therapy. Other considerations include azathioprine, mycophenolate mofetil, or methotrexate. A recent small-scale open-label study showed a reduction in relapse using eculizumab, an inhibitor of C5.1
Conclusion
This is a case of a patient with well-controlled SLE who presented with rapidly progressive vision loss secondary to NMOSD. Although rare, this disease has a greater propensity to affect patients with coexisting autoimmune disease. Therefore, new visual symptoms in such patients should prompt diagnostic consideration of NMOSD.
 Atul Kapila, MD, is a rheumatology fellow at Duke University Medical Center, Durham, N.C. He is a graduate of Vanderbilt University Medical Center Internal Medicine Residency, Nashville.
Atul Kapila, MD, is a rheumatology fellow at Duke University Medical Center, Durham, N.C. He is a graduate of Vanderbilt University Medical Center Internal Medicine Residency, Nashville.
 Tayseer Haroun, MBBS, is a rheumatology fellow at Duke University Medical Center, Durham, N.C. She is a graduate of Texas Tech University Health Sciences Center Internal Medicine Residency, Amarillo, Texas.
Tayseer Haroun, MBBS, is a rheumatology fellow at Duke University Medical Center, Durham, N.C. She is a graduate of Texas Tech University Health Sciences Center Internal Medicine Residency, Amarillo, Texas.
 Jayanth Doss, MD, MPH, is an assistant professor of medicine at Duke University in the division of Rheumatology, Durham, N.C.
Jayanth Doss, MD, MPH, is an assistant professor of medicine at Duke University in the division of Rheumatology, Durham, N.C.
References
- Jarius S, Wildemann B, Paul F. Neuromyelitis optica: Clinical features, immunopathogenesis, and treatment. Clin Exp Immunol. 2014 May;176(2):149–164.
- Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015 Jul 14;85(2):177–189.
- Pittock SJ, Lennon VA, de Seze J, et al. Neuromyelitis optica and non-organ-specific autoimmunity. Arch Neurol. 2008 Jan;65(1):78–83.
- Wingerchuk DM, Weinshenker BG. The emerging relationship between neuromyelitis optica and systemic rheumatologic autoimmune disease. Mult Scler. 2012 Jan;18(1):5–10.
- Alexopoulos H, Kampylafka EI, Fouka P, et al. Anti-aquaporin-4 autoantibodies in systemic lupus erythematosus persist for years and induce astrocytic cytotoxicity but not CNS disease. J Neuroimmunol. 2015 Dec 15;289:8–11.