Her basic laboratory tests were unremarkable except for an anemia with a hemoglobin of 11.9 (reference 12.0–16.0 g/dL). A high-sensitivity C-reactive protein (hs-CRP) was 19 (reference <7.4 mg/L) with an erythrocyte sedimentation rate (ESR) of 5 (reference <25 mm/hr). A transesophageal echocardiogram revealed thickening of the ascending aorta, severe aortic insufficiency with a flail right cusp, aortic root dilation and thickening of the valve. Her left ventricular ejection fraction was 35%.
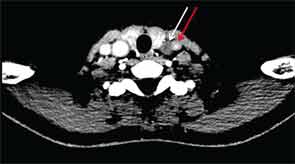
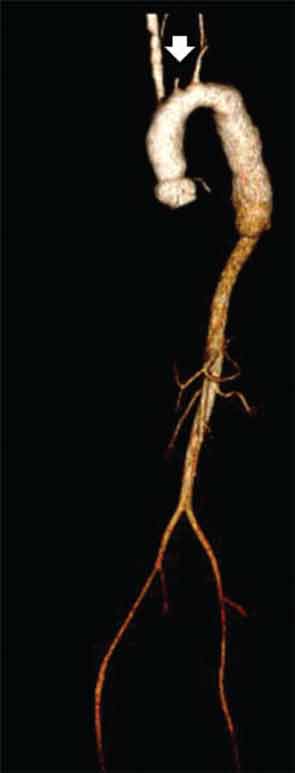
CT of the neck showed complete occlusion of left common carotid artery immediately above the stump (see Figure 1) and complete occlusion of the left internal carotid artery with retrograde filling via the circle of Willis. Significant wall thickening and multiple areas of irregularity and dilatation were seen in the right innominate, right common carotid and left subclavian arteries. A CT angiogram (CTA) of the chest, abdomen and pelvis demonstrated a dilated ascending and descending thoracic aorta with a maximum dimension of 4 cm with concentric wall thickening extending to the midthoracic aorta (see Figure 2). There was no involvement of her abdominal aorta.
The patient was given a pulse dose of glucocorticoids with methylprednisolone 500 mg intravenous daily for three days with improvement in the strength of her left brachial and radial pulses over the course of treatment. She was also initiated on aspirin 81 mg daily and azathioprine 50 mg daily, which was further titrated as an outpatient. After the glucocorticoid pulse, the dose was reduced to prednisone 1 mg/kg/day. Aortic valve replacement was deferred in order to treat the inflammatory vascular process and because she was well compensated clinically. She returned a month later and continued to have stable heart failure symptoms, although she had episodes of left-side amaurosis fugax, lasting less than 15 seconds. Her glucocorticoid dose will be tapered in anticipation of an aortic valve replacement.
Case 2
A 31-year-old Hispanic woman presented to the hospital with hypertension and renal failure.
At age 28, she presented with hypertensive urgency and intestinal angina and was found to have renal artery stenosis. Magnetic resonance angiography (MRA) at that time revealed evidence of a large-vessel vasculitis of the abdominal aorta near the renal arteries, as well as the superior mesenteric artery, and she was diagnosed with Takayasu’s arteritis.
She was treated with high-dose glucocorticoids, azathioprine and infliximab infusions of 5 mg/kg every eight weeks. During tapering of her prednisone dose, she developed abdominal pain suggestive of intestinal ischemia. The dose and frequency of infliximab infusions were increased to 5 mg/kg every six weeks. On this regimen, she was able to taper off prednisone and then, four months prior to admission, to taper off azathioprine.
One month prior to admission she again developed postprandial abdominal pain, which improved after a short course of prednisone. The week prior to admission, the patient developed worsening headache and frothy urine, and she was found to have a blood pressure of 175/104 mmHg in the right arm at her infusion visit. She denied fevers, chills, weight loss, abdominal pain, arm or leg claudication, rash or other new symptoms.
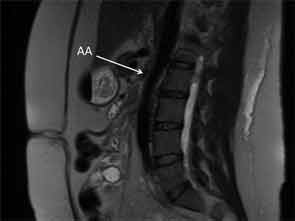
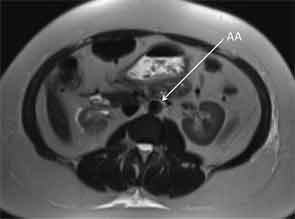
On admission to the hospital, her blood pressure was 200/108 mmHg with no remarkable differences between all four extremities. ESR was 35 mm/hr, compared to a baseline of 7 mm/hr. Her complete blood count was unremarkable. Potassium was 5.1 mmol/L with a creatinine of 3.95 mg/dL. Urinalysis revealed an absence of red and white blood cells, and a urine protein to creatinine ratio was 0.47. Renin activity and aldosterone were both elevated at 52 pg/mL and 46.6 ng/dL respectively with a normal ratio, suggestive of renovascular disease. Duplex ultrasound was a limited study showing decreased flow in the right kidney, which appeared atrophic. Flow velocities were elevated in the left renal artery. An MRA without gadolinium revealed wall thickening of the abdominal aorta most prominent at the level of the superior mesenteric artery (SMA) extending to below the location of the renal arteries with wall thickening of the proximal SMA and left renal artery (see Figure 3). The right renal artery was diminutive with atrophy of the right kidney, similar to studies in 2011.

Lisinopril was discontinued, and 1 g of methylprednisolone was administered intravenously for three consecutive days. Her creatinine rose to a peak of
5.3 mg/dL before improving rapidly over the next several days to 1.4 mg/dL. Blood pressure was controlled with amlodipine and labetalol. The patient was discharged on prednisone 1 mg/kg/day. Infliximab infusions were resumed at 7 mg/kg every four weeks, and azathioprine was reinstituted at 150 mg/day.
Discussion
Takayasu’s arteritis (TAK) is a well-established but rare form of large-vessel vasculitis. It has also been known as pulseless disease, aortic arch syndrome or occlusive thromboaortopathy. It is a chronic inflammatory arteritis affecting large vessels, predominantly the aorta and its main branches. Inflammation leads to thickening of the arterial wall, resulting in occlusion, stenosis, dilatation and thrombus formation. The vast majority of lesions in TAK are stenotic, but more acute inflammation may destroy the arterial media and lead to aneurysm formation, which is seen in up to one-third of TAK patients.1,2
Published descriptions of this form of arteritis date back to 1830. However, the disease received its name from Mikito Takayasu, a professor of ophthalmology, who in 1905 presented the case of a 21-year-old woman with characteristic fundal arteriovenous anastomoses, which later was suggested to result from retinal ischemia. At disease presentation or during relapses, TAK patients may present with nonspecific inflammatory complaints, such as fever, malaise, anorexia, weight loss, myalgia or arthralgias. This can be associated with vascular pain (e.g., carotidynia). As arterial lesions develop, features like decreased or absent peripheral pulses, vascular bruits, discrepancies in blood pressure between limbs, limb claudication, and hypertension arise. Heart failure can also be seen as a result of aortic regurgitation, longstanding hypertension, or coronary artery disease. Transient ischemic attacks, strokes, mesenteric ischemia, and retinopathy are other manifestations of the resultant ischemia. TAK can affect medium-size vessels, such as coronary arteries, and pulmonary vasculopathy and hypertension are also seen in some patients. Extravascular manifestations that have been reported include dermatologic manifestations, such as erythema nodosum and erythema induratum, glomerular lesions and cardiac manifestations, such as dilated cardiomyopathy and myocarditis.3
Takayasu’s arteritis is most commonly seen in Japan, South East Asia, India and Mexico. It is rare in North America, with an estimated incidence of 2.6 per million per year in Olmsted County, Minn.4 Although geographical differences regarding the distribution of arterial lesions have been described in TAK, the aorta is the most affected artery, followed by the subclavian, common carotid and renal arteries.5 The disease commonly presents in the second or third decade of life and predominantly affects women. From the onset of first symptoms, there is often a delay in diagnosis of months to years.
The American College of Rheumatology has established classification criteria for TAK (see Table 1), which were primarily designed to distinguish this disorder from other forms of vasculitis for study purposes. This classification system has a sensitivity and specificity of 90.5% and 97.8%, respectively.6 Attempts have been made to classify the disease on the basis of angiographic findings (see Figure 4), which can be useful in allowing a comparison of patient characteristics according to the involved vessels and helpful in planning surgery.7 However, this provides little information on prognosis.
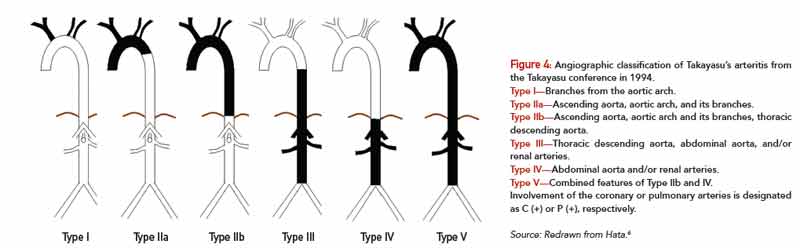
Type I—Branches from the aortic arch.
Type IIa—Ascending aorta, aortic arch, and its branches.
Type IIb—Ascending aorta, aortic arch and its branches, thoracic descending aorta.
Type III—Thoracic descending aorta, abdominal aorta, and/or renal arteries.
Type IV—Abdominal aorta and/or renal arteries.
Type V—Combined features of Type IIb and IV.
Involvement of the coronary or pulmonary arteries is designated as C (+) or P (+), respectively.
Source: Redrawn from Hata.6
Conventional angiography remains the gold standard for the diagnosis of TAK. MRA and CTA can also be used to evaluate large- and medium-size vessels. Rheumatic diseases that affect the aorta or its branches include giant cell arteritis, primarily differentiated from Takayasu’s by age of disease onset and distribution of lesions. Granulomatous vasculitis can be demonstrated on histologic examination in both diseases. Other rheumatic diseases, such as relapsing polychondritis, ankylosing spondylitis, Behçet’s disease and Cogan’s syndrome, may have a similar arteriographic appearance to TAK but can be differentiated on the basis of their other clinical manifestations. Infectious or “mycotic” aneurysms, atherosclerotic disease, fibromuscular dysplasia, congenital co-arctation and inherited disorders of connective tissue, such as Marfan’s syndrome and Ehlers Danlos, must also be considered.
The mainstay of therapy is suppression of inflammation and preservation of vascular competence. Initial treatment is typically glucocorticoids at a dose of 1 mg/kg/day. Although the majority of patients will respond to this treatment, some cases are glucocorticoid resistant. In addition, relapse with tapering steroids is common. Most patients will require other immunosuppressive agents, which are often initiated at the time of diagnosis.8 An initial open-label study of methotrexate with glucocorticoids in 16 patients showed remission in 81% of patients and sustained remission in 50% of patients, suggesting its utility as a steroid-sparing agent.9 Limited data also exists to support the use of azathioprine, mycophenolate, cyclosporine, tacrolimus, leflunomide or cyclophosphamide.10 In all cases, a significant number of patients continue to have active disease or are unable to discontinue glucocorticoid treatment.
The granulomatous nature of inflammation in Takayasu’s arteritis suggests a potential role of tumor necrosis factor-alpha in disease pathogenesis. Hoffman et al published the first open-label prospective study in 2004 of 15 patients treated with infliximab or etanercept. Most patients had previously received other immunosuppressive agents in addition to glucocorticoids. Response was seen in 14 of 15 patients, with 10 patients experiencing sustained remission for one to 3.3 years off glucocorticoid therapy.11 Since that time, reviews of published case series note similar results with good response rates and the ability to taper prednisone. The majority of patients have been treated with traditional immunosuppressants, such as methotrexate or azathioprine, in addition to biologic therapy, most commonly infliximab. Relapse is common, however, as is the need for dose escalation of anti-TNF therapy.12,13
More recently, anti-interleukin-6 therapy with tocilizumab has been reported in case reports and small case-series of patients refractory to other treatments including, in some cases, anti-TNF therapy. Although results have been encouraging, it’s important to note the small numbers, limited follow-up and lack of controlled or even prospective trials to date. Even less data exists to guide the use of rituximab, although this remains an active area of investigation.12 The ongoing phase II AGATA trial (Abatacept for Treating Adults with Giant Cell Arteritis and Takayasu’s Arteritis) is the first randomized controlled trial in TAK. Additional controlled trials are needed to guide optimal therapy. Additionally, the best strategy to assess response to therapy is unknown but includes assessing symptoms, monitoring inflammatory markers and performing interval imaging to identify new or progressive vascular lesions.
Although immunosuppressant therapy may treat inflammation and prevent new vascular lesions, surgical or endovascular interventions are sometimes required to treat chronic, severe vascular complications. These include bypass grafting, endarterectomy, aortic valve replacement, angioplasty and stenting. Indications for intervention include extremity ischemia limiting activities of daily living, cerebral ischemia or symptomatic stenosis > 70% of cerebral vessels, refractory hypertension due to renal artery stenosis, coronary artery stenosis causing ischemia, moderate to severe aortic regurgitation, severe aortic co-arctation and progressively enlarging or dissecting aneurysms.11 Interventions should be avoided if possible and should only be performed when severe complications are present.
Procedures in patients without major complications have been associated with decreased survival.14,15 A higher rate of restenosis or occlusion after grafting, angioplasty or stenting has been observed in patients with TAK compared with patients with atherosclerotic disease. This difference may be due to the persistent inflammation observed in some operative specimens in patients thought to be in remission or because of reduced compliance of fibrotic vessels.5,16 The literature suggests that surgery is associated with a somewhat lower rate of complications than endovascular intervention. Importantly, outcomes are improved if procedures are done after active inflammation is treated.15-17 Whenever possible, medical treatment should come before surgical intervention.
In the first case, Takayasu’s arteritis involved the aortic arch causing amaurosis fugax and symptoms from aortic regurgitation. The active inflammation of her Takayasu’s arteritis was treated medically before proceeding with surgical treatment of her severe aortic regurgitation.
In the second case, Takayasu’s arteritis primarily involved the abdominal aorta and renal vasculature. Her disease stabilized on combination therapy with azathioprine and infliximab, but recurred with tapering of therapy. Active involvement of the left renal artery at presentation caused hypertension and acute renal failure. Her renal function improved with immunosuppressive treatment and her hypertension was controlled with medical therapy and without the need for endovascular intervention.
Conclusion
These cases demonstrate some of the various manifestations of TAK as it affects different portions and branches of the aorta. Prompt diagnosis requires a knowledge of the potential clinical manifestations of large-vessel vasculitis. Optimal treatment of this complicated disease remains an area of active investigation. It requires close patient monitoring, trials of various immunosuppressive regimens and, at times, surgical or endovascular intervention.
Ashwini Komarla, MD, is a fellow in the Division of Rheumatology at the University of Pennsylvania in Philadelphia.
Michael George, MD, is also a fellow in the Division of Rheumatology at the University of Pennsylvania in Philadelphia.
Antoine Sreih, MD, is assistant professor at Penn Vasculitis Center, Division of Rheumatology at the University of Pennsylvania in Philadelphia.
Chris Derk, MD, MS, is associate professor and program director in the Division of Rheumatology at the University of Pennsylvania in Philadelphia.
References
- Maksimowicz-McKinnon K, Clark TM, Hoffman GS. Limitations of therapy and a guarded prognosis in an American cohort of Takayasu arteritis patients. Arthritis Rheum. 2007;56: 1000–1009.
- Mason JC. Takayasu arteritis—advances in diagnosis and management. Nat Rev Rheumatol. 2010; 6:406–415.
- Sharma BK, Jain S, Sahar S. Systemic manifestations of Takayasu arteritis: The expanding spectrum. Int J Cardiol. 1996;54(2):S149–S154.
- Hall S, Barr W, Lie JT, et al. Takayasu arteritis: A study of 32 North American patients. Medicine (Baltimore). 1985;64(2):89–99.
- Kerr GS, Hallahan CW, Giordano J, et al. Takayasu Arteritis. Ann Intern Med. 1994;120(11):919–929.
- Arend WP, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum. 1990;33(8):1129–1134.
- Hata A, Noda M, Moriwaki R, et al. Angiographic findings of Takayasu arteritis: New classification. Int J Cardiol. 1996;54:S155–S163.
- Kötter I, Henes JC, Wagner AD, et al. Does glucocorticosteroid-resistant large-vessel vasculitis (giant cell arteritis and Takayasu arteritis) exist and how can remission be achieved? A critical review of the literature. Clin. Exp. Rheumatol. 2012;30:S114–S129.
- Hoffman GS, Leavitt RY, Kerr GS, et al. Treatment of glucocorticoid-resistant or relapsing Takayasu arteritis with methotrexate. Arthritis Rheum. 1994;37(4):578–582.
- Keser G, Direskeneli H, Aksu K. Management of Takayasu arteritis: A systematic review. Rheumatology (Oxford). 2013. doi:10.1093/rheumatology/ket320.
- Hoffman GS, Merkel PA, Brasington RD, et al. Anti-tumor necrosis factor therapy in patients with difficult to treat Takayasu arteritis. Arthritis Rheum. 2004;50(7):2296–2304.
- Clifford A, Hoffman GS. Recent advances in the medical management of Takayasu arteritis: An update on use of biologic therapies. Curr Opin Rheumatol. 2014;26(1):7–15.
- Comarmond, C. Plaisier E, Dahan K, et al. Anti TNF-α in refractory Takayasu’s arteritis: Case series and review of the literature. Autoimmun. Rev. 2012;11(9):678–684.
- Miyata T, Sato O, Koyama H, et al. Long-term survival after surgical treatment of patients with Takayasu’s arteritis. Circulation. 2003;108(12):1474–1480.
- Perera AH. Youngstein T, Gibbs RG, et al. Optimizing the outcome of vascular intervention for Takayasu arteritis. Br J Surg. 2014;101(2):43–50.
- Liang P, Tan-Ong M, Hoffman GS. Takayasu’s arteritis: Vascular interventions and outcomes. J Rheumatol. 2004;31(1):102–106.
- Saadoun D, Lambert M, Mirault T. Retrospective analysis of surgery versus endovascular intervention in Takayasu arteritis: A multicenter experience. Circulation. 2012;125(6):813–819.