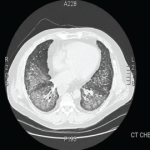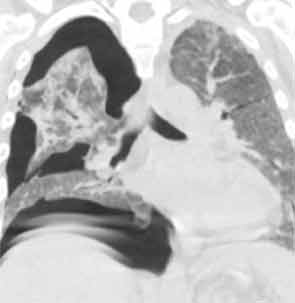
His tracheostomy was decannulated three weeks later. He required pleurodesis for recurrent pneumothorax, but by mid February, the last chest tube was removed. At the time of his discharge to a rehabilitation facility, his oxygen saturation was 95–97% on room air.
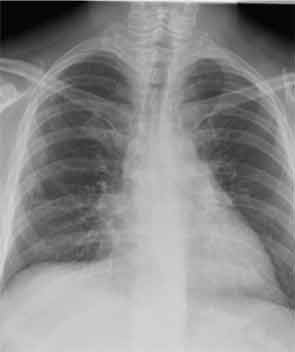
He has since been slowly recovering from critical-illness myopathy. In early March, he was discharged home. Prednisone has been tapered in 10-mg decrements monthly. At the end of March, we saw him back in our outpatient clinic for the first time. He had regained about 30 pounds since hospital discharge. He had nearly full strength in all extremities. He remained B-cell depleted and continued to have a remarkable sustained response to steroids and rituximab in terms of his lung disease. Figure 5 (below) shows his most recent chest X-ray.