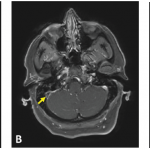
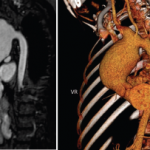
Along the way, we have discovered possible differences between patients with Behçet’s in the U.S. and patients from areas where BS is more endemic. We also have found that this syndrome may not be as rare in the U.S. as was once assumed, especially in the New York area, where the ethnic and racial mix may contribute to the increased incidence of BS.
Clinical Features
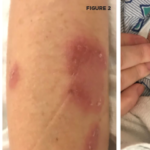
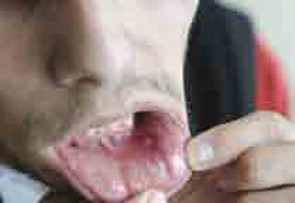
Behçet’s syndrome is a systemic vasculitis affecting the small and large vessels of both the venous and arterial systems and has an unknown etiology. It was first described by Hulusi Behçet in 1937 on the basis of three patients with a triple-symptom complex of oral aphthae, genital ulcers, and hypopyon uveitis.1 Subsequent studies showed that this entity was a multisystem disease characterized by variable clinical manifestations. Virtually all patients have recurrent oral aphthae (see Figure 1, below), with successively fewer having genital ulcers, skin lesions, arthritis, uveitis, thrombophlebitis, and gastrointestinal (GI) and central nervous system (CNS) involvement.