
During the past five years, an explosion of information has emerged from genetic association studies of systemic lupus erythematosus (SLE). The sheer number of genes identified through this work, as well as the specific genes that have been identified as SLE risk factors, demonstrate the surprising degree of success of genome-wide association and related studies to reveal the genetic underpinnings of this complex disorder. Further, the remarkable degree of clustering of recently identified variants into specific biologic pathways has, to a greater extent than anticipated, provided insights into the etiologic processes that contribute to SLE. Also surprising has been the extent to which recently identified genetic variants appear to be relevant to multiple autoimmune diseases, highlighting the close etiologic relationships among this broad and clinically diverse group of human disorders. Given the rapid pace of gene discovery in SLE, it is helpful to review the relevant concepts and findings of this research, including what they reveal about this complex disorder. In addition, I also will briefly describe in this article the direction of future research in this area.
A Genetically Complex Disorder
Although the prevalence of SLE is relatively low, recent genetic discoveries highlight the close etiologic relationships among the large and diverse group of human autoimmune disorders. These findings are consistent with the well-known clustering of these diseases within families and individuals. The genetic complexity of SLE means that the disease results from the action of many predisposing genes as well as environmental and other nongenetic factors. As a consequence of this complexity, SLE does not demonstrate classical Mendelian modes of familial inheritance, and there is a lack of correspondence between the genotype that one inherits and disease status. Complex genetic disorders also are characterized by heterogeneity of genetic associations, according to ethnic and clinical differences, for example; this heterogeneity has been strongly reinforced by recent genetic studies of SLE.

In spite of these complexities, there is clear evidence of familial clustering for SLE, supporting an important role for genetics. For example, siblings of SLE patients are approximately 30 times more likely to develop SLE compared with individuals without an affected sibling. This ratio is referred to as ls and is notably higher in SLE compared with many other autoimmune diseases. For example, the ls for psoriasis and rheumatoid arthritis is in the range of 5–10. Although the relatively larger ls for SLE should theoretically translate into a greater ease of identifying SLE genes, it is important to remember that this parameter reflects the action of all genetic risk factors. Thus, the ease with which SLE genes can be identified will vary greatly depending upon whether the increased risk of disease in families is due to the action of 30 genes versus 300 or even 3,000 genes.
Genome-Wide Association Studies
The pace of gene discovery in SLE has increased exponentially during the past five years due primarily to the increased feasibility of performing very large genome-wide association studies (GWAS) using hundreds of thousands of single-nucleotide polymorphism (SNP) markers. (See Definitions (at right) for a brief definition of SNP and other technical terms used here.) The first GWAS of a complex human disease was completed in 2006, and since then, application of this methodology to SLE has resulted in the discovery of over 20 firmly established risk loci. This rapidity of recent progress is in contrast to the previous slow pace of gene discovery that began in the 1970s with the discovery of HLA gene associations with SLE and was followed by additional discoveries that largely paralleled developments in molecular genetic technologies.1
During the past two years, four GWAS of SLE cases and controls of European descent have been published, followed by two GWAS of SLE cases and controls of Asian ancestry published more recently.2–7 Figure 1 (above) summarizes the results from one of these studies and provides a graphical representation of the key features of these study designs.2 In brief, each dot in this figure (known as a Manhattan plot due to the similarity to the Manhattan skyline) corresponds to a genetic marker that, in this particular study, included ~550,000 SNPs. The dots are color coded and arranged along the x-axis according to position (with each color representing a different chromosome). The y-axis represents the significance level (–log P value) for the association of each SNP with SLE (i.e., comparison between SLE cases and controls). Given the size of this study, with more than 500,000 SNPs, the multiple testing burden is quite high and therefore the level of significance for definitive genetic associations is considered to be approximately 5 x 10–8, with results falling between –log P values of approximately 5–7.3 representing associations of borderline significance. The latter findings have been the intense focus of follow-up investigations, as described below. Thus, on the basis of this single experiment, which included about 1,300 SLE cases and approximately 3,300 control individuals of European ancestry, the HLA, STAT4, and IRF5 loci exceeded the level of significance for definitive results, and the top novel loci were BLK and ITGAM-ITGAX, which exceeded this threshold following genotyping in additional SLE cases and controls of European ancestry.
More recently, a large group of investigators from the United States and Sweden performed a follow-up study of the top loci from the aforementioned GWAS to identify additional risk loci.8 More specifically, the investigative team genotyped more than 3,000 SNPs from approximately 2,500 distinct regions that showed nominal evidence of association with SLE (P < 0.05) in an independent sample of about 2,000 SLE cases and about 4,300 controls. This replication effort identified five new SLE susceptibility loci (with p < 5 x 10–8) in TNIP1, PRDM1, JAZF1, UHRF1BP1, and IL10. Of interest, but not unexpected for a genetically complex disease, the strength of association for these loci was modest, with odds ratios ranging from 1.17 (for UHRF1BP1) to 1.27 (for TNIP1). Also as expected based on characteristics of the markers chosen for genotyping, the associated variants were relatively common, with minor allele frequencies greater than 5%. This study also identified 21 candidate loci with P ≤ 1 x 10–5, which will undoubtedly be the focus of additional studies. Lastly, these authors analyzed alleles previously associated with other autoimmune diseases and found evidence to support association with SLE for five additional loci (P < 1 x 10–3), including IFIH1, CFB, CLEC16A, IL12B, and SH2B3. These results, together with the prior GWAS and other recent studies have expanded the number of established SLE susceptibility loci to approximately 30.9 Importantly, these recent findings implicate several key immunologic pathways in SLE pathogenesis on the basis of the clustering of these genes into specific biologic pathways, including those related to immune complex processing, immune signal transduction, and the toll-like receptor (TLR) and type 1 interferon (IFN-1) pathways (see Figure 2, p. 31).10
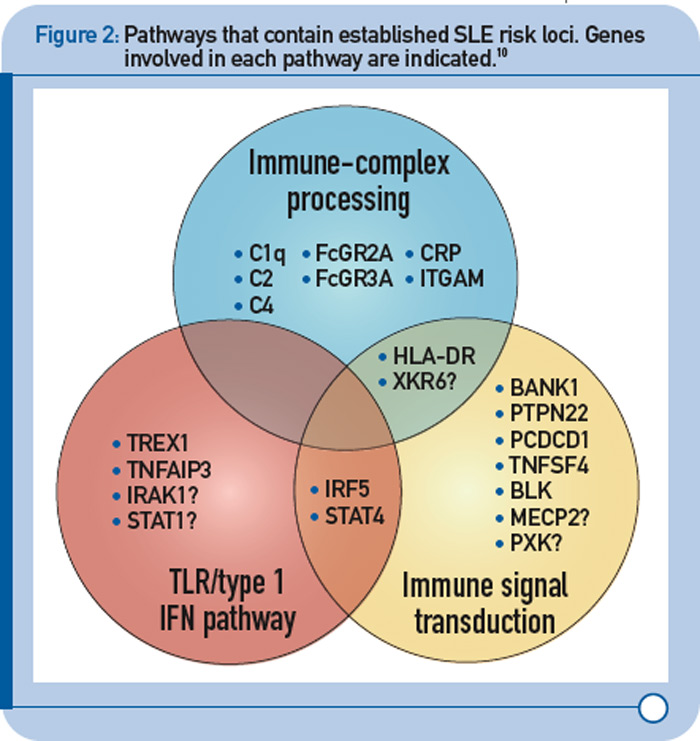
TLR and IFN-1 Pathways
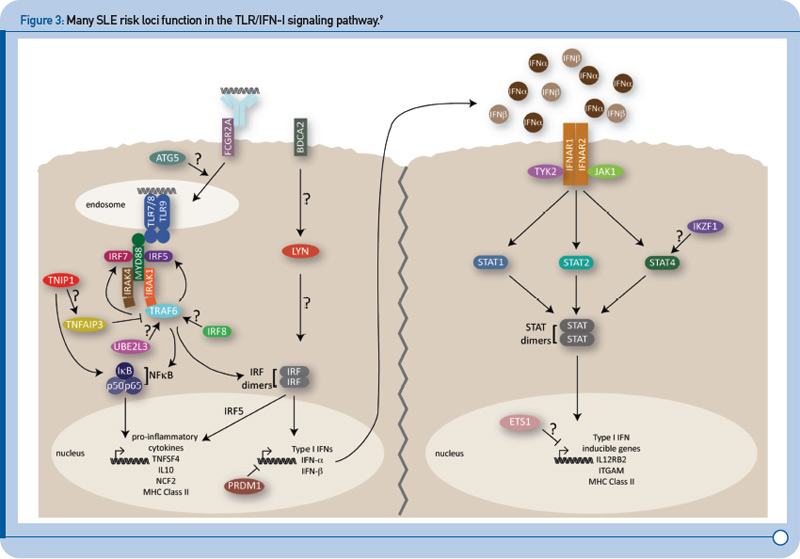
Recent genetic discoveries highlight importance of these two pathways in SLE. Perhaps most striking has been the extent to which recent discoveries in SLE highlight the importance of the TLR and IFN-1 signaling pathways.9 For example, Figure 3 (p. 31) shows key components of these interrelated pathways, many of which have now been firmly established as SLE risk loci based on recent genetic studies.
One of the key components of these pathways, TNFAIP3, nicely illustrates the extent to which some of the recently identified risk loci are clearly relevant to multiple autoimmune disorders. TNFAIP3, which was initially implicated in an early GWAS of Crohn’s disease, has now been implicated in genetic studies of at least six autoimmune disorders, including SLE. Thus, this gene may represent the best example of a true autoimmunity locus with relevance to a broad spectrum of disease, outside the HLA complex of genes. Many questions remain, however, about the mechanism by which genetic variation in the TNFAIP3 region contributes to autoimmunity. Illustrative of these challenges is the fact that many different variants, which span a broad genomic region, have been implicated with little overlap across diseases in the most strongly associated variants. Further, although some studies have implicated coding and intronic SNPs that may be functionally relevant, much work remains to establish the specific mechanisms by which these or other variants contribute to autoimmune disease processes. Nonetheless, compelling functional data exists for some of the recently identified SLE risk loci. Table 1 (p. 32) lists some of the genes for which evidence supporting a functional role of the associated variants has been reported.
Genetics and Disease Heterogeneity
In addition to the insights that findings related to TNFAIP3 and other genes have provided about fundamental autoimmune processes, recent work also highlights the fact that many recently established risk loci may predispose more specifically to certain types of disease. For example, following the identification of STAT4 as a genetic risk factor for rheumatoid arthritis and SLE, additional work demonstrated that the implicated STAT4 variant is associated more specifically with severe forms of SLE.11 For example, compared to the frequency of the risk variant in control individuals of 22.5%, the frequency of the risk variant was about 30% among individuals with disease characterized by oral ulcers and/or photosensitivity (odds ratio [OR] ~ 1.5), 35% among patients whose disease is characterized by anti-dsDNA autoantibody production (OR ~ 1.9), and 38% among patients with severe renal involvement (OR > 2.0). Similarly, work underway now suggests that most of the recently identified SLE risk variants are more strongly associated with anti-dsDNA positive disease compared with anti-dsDNA negative disease.12 Thus, an important area for future work will be to further refine genotype–phenotype associations in SLE in order to dissect, with greater etiologic relevance, this genetically, and clinically complex disease.
Current Status and Future Directions
In spite of the success of recent GWAS and other genetic studies, at least in terms of the number and specific loci identified, it has been surprising to realize that these loci, collectively, explain only a minority of the heritability of the disease. However, the same can be said about most complex diseases that have been the focus of GWAS and related studies over the past five years.13 Thus, an important question that is the focus of much current investigation in this area is what explains this “missing heritability.” There are a number of possibilities, many of which have already received some support.
For example, it is possible that a very large number of additional variants, with increasingly smaller association strength, are contributing to disease susceptibility, in which case even larger studies may be required to identify these loci. Another possibility is that much of the missing heritability reflects the action of a large number of rare variants that have not been specifically targeted in recent GWAS and other genetic studies. These variants will require different approaches for identification, such as high-throughput DNA sequencing. Another category of genetic variation that has not been specifically targeted or captured in most recent genetic studies are structural variants, such as copy-number variation, as has been previously demonstrated for C4 and FcgR genes in SLE.

Epigenetics and the Environment
Recent data also provide compelling support for a role of epigenetic factors in SLE. Epigenetics refers to inherited changes in gene expression caused by mechanisms other than DNA base sequence changes. The most well understood type of epigenetic factor is DNA methylation, which has been shown to play a role in a variety of human processes, such as X chromosome inactivation and certain cancers. Recent work by Javierre et al suggests that differences in the DNA methylation status of genes may explain, at least in part, the discordance observed in some identical twins that are discordant for SLE.14 More specifically, they found that twins with SLE were characterized by lower levels of DNA methylation overall, including approximately 50 genes with established roles in immune responses, cytokine production, and cell activation. Previous research has also implicated the importance of DNA methylation in SLE. Given the strong impact of environmental factors on the DNA methylation status of genes, as strikingly illustrated by the Agouti mouse model in which changes in the methylation content of the diet have profound effects on the coat color and other phenotypic characteristics (e.g., obesity, diabetes), epigenetic mechanisms could potentially provide a missing link between genetic and environmental risk factors for SLE. As with many of the aforementioned unanswered questions, epigenetics will likely be an intense focus of future investigation.
The extent to which recently identified SLE susceptibility loci appear to be relevant to other autoimmune disorders, and vice versa, has reinforced the importance of family history in the evaluation of SLE patients.

Although much work remains to be done to fully define the genes and epigenetic factors that contribute to disease risk and outcome in SLE, it is interesting to consider how this information may contribute to clinical practice in the future. The extent to which recent genetics studies have informed our understanding of disease mechanisms in SLE is impressive, and our knowledge will continue to develop as we more fully elucidate the genetic underpinnings of this and related disorders. Such information has great potential to accelerate the identification of targets for novel therapies, as reviewed recently by Plenge and Raychaudhuri.15 One can also envision the incorporation of genetic information into a new generation of diagnostic or classification criteria, which would be informed by knowledge about specific disease subsets that are more homogeneous in terms of disease etiology, prognosis, and treatment response. Certainly, the extent to which recently identified SLE susceptibility loci appear to be relevant to other autoimmune disorders, and vice versa, has reinforced the importance of family history in the evaluation of SLE patients. It is clear that a family history of a wide range of autoimmune diseases may have relevance for an individual patient being evaluated for a possible diagnosis of SLE.
Lastly, the ultimate goal of this and other etiologic studies is to enable early intervention and disease cure or prevention. And while that may seem like an unlikely scenario, only the most optimistic among us imagined 10 years ago that in 2011 we would be talking about 30 genes of proven relevance to SLE!
Dr. Criswell is a professor of medicine and orofacial sciences at the University of California, San Francisco.
References
- Moser KL, Kelly JA, Lessard CJ, Harley JB. Recent insights into the genetic basis of systemic lupus erythematosus. Genes Immun. 2009;10:373-379.
- Hom G, Graham RR, Modrek B, et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med. 2008;358:900-909.
- Harley JB, Alarcon-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204-210
- Graham RR, Cotsapas C, Davies L, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet. 2008;40:1059-1061.
- Kozyrev SV, Abelson AK, Wojcik J, et al. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet. 2008;40:211-216.
- Han JW, Zheng HF, Cui Y, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet. 2009;41:1234-1237.
- Yang W, Shen N, Ye DQ, et al. Genome-wide association study in Asian populations identifies variants in ETS1 and WDFY4 associated with systemic lupus erythematosus. PLoS Genet. 2010;6:e1000841.
- Gateva V, Sandling J, Hom G, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1, and IL10 as novel risk loci for systemic lupus erythematosus. Nat Genet. 2009;41:1228-1233.
- Flesher DL, Sun X, Behrens TW, Graham RR, Criswell LA. Recent advances in the genetics of systemic lupus erythematosus. Expert Rev Clin Immunol. 2010;6:461-479. PMCID: 2897739.
- Harley IT, Kaufman KM, Langefeld CD, Harley JB, Kelly JA. Genetic susceptibility to SLE: New insights from fine mapping and genome-wide association studies. Nat Rev Genet. 2009;10:285-290.
- Taylor KE, Remmers EF, Lee AT, et al. Specificity of the STAT4 genetic association for severe disease manifestations of systemic lupus erythematosus. PLoS Genet. 2008;4:e1000084.
- Chung SA, Taylor KE, Graham RR, et al. Differential genetic associations for systemic lupus erythematosus based on anti-dsDNA autoantibody production. PLoS Genetics. 2011: in press.
- Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747-753. PMCID: 2831613.
- Javierre BM, Fernandez AF, Richter J, et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010;20:170-179.
- Plenge RM, Raychaudhuri S. Leveraging human genetics to develop future therapeutic strategies in rheumatoid arthritis. Rheum Dis Clin North Am. 2010;36:259-270. PMCID: 2879392.
- Graham RR, Kyogoku C, Sigurdsson S, et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc Natl Acad Sci U S A. 2007;104:6758-6763.
- Bottini N, Musumeci L, Alonso A, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet. 2004;36:337-338.
- Nath SK, Han S, Kim-Howard X, et al. A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet. 2008;40:152-154.
- Musone SL, Taylor KE, Lu TT, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet. 2008;40:1062-1064.

