
Gout is an inflammatory arthritis associated with hyperuricemia. According to recent data, the prevalence of gout and hyperuricemia in U.S. adults has been increasing over the past 20 years, with current estimates of 3.9% and 21%, respectively.1 New research has put focus on the interplay of hyperuricemia, gout and metabolism. Patients with gout and hyperuricemia contend with a high prevalence of comorbidities, including hypertension, renal disease, obesity, diabetes mellitus (DM) and cardiovascular disease, as well as the metabolic syndrome (MetS).2 In this review, we focus on the association of hyperuricemia and gout with metabolic derangement, including the MetS, DM and obesity.
Metabolic Syndrome
The MetS, as defined by the 2001 Adult Treatment Panel III criteria, includes abdominal obesity, hypertriglyceridemia, low high-density lipoprotein (HDL) cholesterol, high blood pressure and high fasting glucose. NHANES data estimate the prevalence of the MetS at a striking 22.9%.3 Debate persists as to whether uric acid (UA) has a causative role in the MetS and should be considered in the guidelines, vs. simply being a marker of metabolic derangement.4-6

On the one hand, a physiologic case can be made for MetS leading to an increase in UA levels. On the other hand, mechanisms for increased UA levels causing MetS have also been described. Elevated levels of insulin increase UA reabsorption at the renal proximal tubule via stimulation of urate-anion exchanger URAT1 and the Na+-dependent anion co-transporter, leading to elevation in serum UA levels.11 Hyperinsulinemia may also diminish oxidative phosphorylation, resulting in elevated adenosine levels, which decrease renal UA excretion and increase UA production.7 Additionally, leptin levels increase with obesity; rising leptin is felt to be independently associated with and contribute to increasing UA levels via reduced renal excretion.12 Alternatively, UA levels could contribute to components of the MetS. Hyperuricemia may cause endothelial dysfunction and a reduction in endothelial nitric oxide (NO).13 NO is integral in promoting glucose influx to skeletal muscle; disruption to the action of insulin could lead to insulin resistance.13 Lastly, UA has been implicated in the proinflammatory milieu of adipose tissue. UA has been found in vitro to increase monocyte chemotactic protein-1 (MCP‑1), a proinflammatory adipokine, as well as decrease adiponectin, a known antiinflammatory and insulin sensitizer.14 Supporting the role of UA contributing to MetS, treatment with allopurinol in hyperuricemic obese mice decreases MCP-1, increases adiponectin, and improves insulin resistance.14
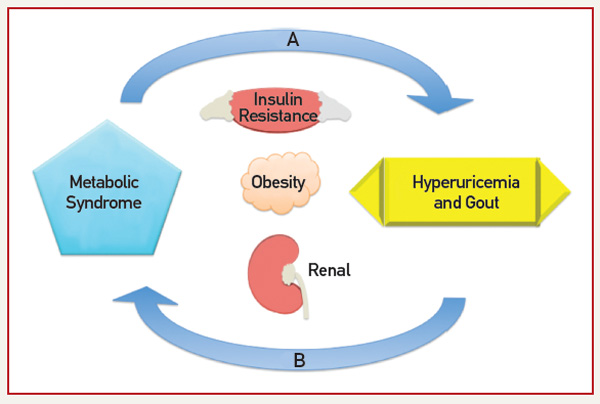
Fructose, found in sugar-sweetened soft drinks, has recently come under fire for its role in promoting hyperuricemia, gout and the MetS.15 Fructose metabolism increases UA through purine nucleotide degradation and de novo purine synthesis, setting the stage for UA-induced NO inhibition.15,16 Indeed, rats on a high-fructose diet treated with allopurinol or benzbromarone benefit from prevention or reversal of MetS components, providing interventional support to the idea that UA may be a cause of MetS.16 Figure 1 (right) demonstrates the bidirectional relationship between UA and the MetS as mediated through insulin resistance, obesity and renal excretion.
Diabetes
As described above, UA is associated with insulin resistance. Data also indicate a link between UA, gout and DM. A retrospective analysis demonstrates the risk of incident DM to be significantly higher in patients with hyperuricemia (UA >7), suggesting that 8.7% cases of new-onset DM are attributable to hyperuricemia.17 Prior prospective data report an even higher estimate of one-quarter of all DM diagnoses attributed to UA.18 Large prospective cohort studies, including the Framingham Heart Study and the Coronary Artery Risk Development in Young Adults, have confirmed UA to be a predictor for DM development.19,20 A cohort study notes a 78% increase in risk of impaired fasting glucose or DM when the highest UA quintile was compared with the lowest.21 Interestingly, although patients with gout appear to have an increased risk of DM, patients with DM tend to be protected from gout. In a cohort of men with high cardiovascular risk, having a diagnosis of gout is associated with a 1.3 times higher risk of incident DM.22 Conversely, a recent case-control study reports decreased incident gout with increasing hemoglobin A1c (HbA1c) values over 7% and DM duration, suggesting a protective role for DM.23
The complicated relationship between UA, gout and DM was described by Choi et al using the cross-sectional NHANES cohort. UA increases with rising HbA1c levels until reaching a level of 6–6.9%, after which UA levels decrease, creating an inverse U-shaped association when UA is plotted against HbA1c.11 UA demonstrates a similar inverse U-shaped curve when graphed against fasting glucose levels; however, UA increases linearly with increasing fasting C-peptide levels and homeostasis model assessment-estimated insulin resistance.11 This finding was notable in two aspects:
- UA levels appeared to be higher in the pre-diabetic phase and lower with overt DM; and
- Insulin (represented by C-peptide and HOMA-IR) and glucose appeared to have separate and independent effects on UA levels.
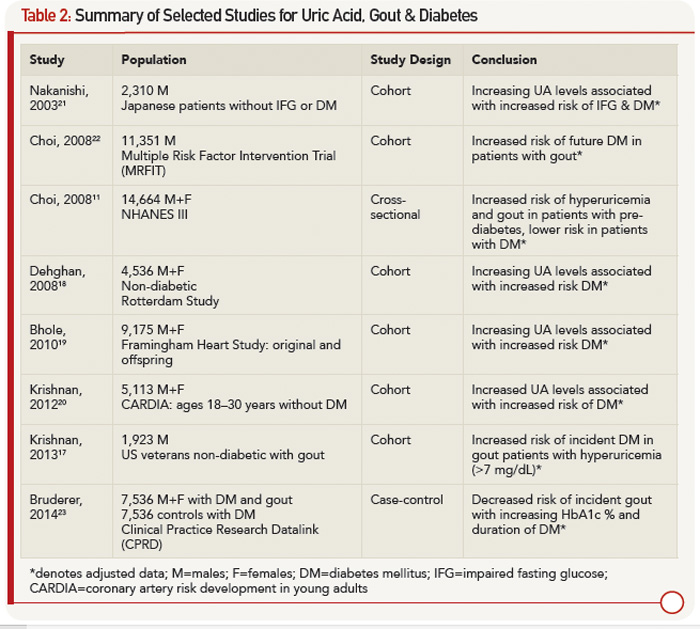
From a clinical standpoint, it appears that patients are at higher risk for gout in the pre-diabetic period, and patients with DM may indeed be protected. Table 2 outlines studies showing an increased risk of DM in patients with elevated UA and gout, as well as studies demonstrating the protective effect of DM for hyperuricemia and gout.
Mechanistically, insulin affects UA reabsorption via the kidney, as outlined above. In comparison, glucose is thought to reduce UA, with glucose levels higher than 180 mg/dL being uricosuric.11 The balance of hyperinsulinemia and hyperglycemia and the competing effect on renal urate clearance may explain the clinical observations. Inflammation is also a well-known component to the development of DM.
As discussed above, UA may have a role as a proinflammatory substrate. Accordingly, new pharmacologic considerations for managing DM include agents targeting inflammation, such as interleukin-1 beta (IL-1β) antagonists. This therapeutic approach is attractive given the success of IL-1β antagonists in the management of gout and the large number of patients with both DM and gout.24 Indeed, a case review of three patients with gout and DM treated with anakinra (IL-1β receptor antagonist) after failing conventional urate lowering and antiinflammatory therapy saw improvement in not only gout manifestations, but they also had decreased fasting glucose and HbA1c levels.25
Obesity
As we come to understand adipose tissue as a metabolically active entity, obesity and weight gain have also been implicated in gout. In fact, higher body mass index (BMI) and obesity have demonstrated the strongest association with hyperuriciemia and gout.26
Cross-sectional data from NHANES show that the prevalence of gout increases with increasing BMI even after adjustment for UA, with the prevalence being 1.3 times greater in overweight individuals and up to 2.2 times greater in the morbidly obese when compared with normal BMI.27 A prospective cohort study in women supports this, finding early- to mid-life obesity an independent risk factor for incident gout.28 Obesity has been associated with earlier onset gout. The age of incident gout decreases by 4.5 years with every 5 kg/m2 increase in BMI at age 21.29 Even in children 9–15 years old, UA levels have been found to be higher in those overweight.30
Visceral adiposity is positively associated with UA levels, potentially due to the compounding effects of leptin, adiponectin and insulin on reducing renal excretion or from triglyceride-induced increased UA production.31
Weight gain itself is a risk factor for incident gout; a cohort study demonstrates that weight gain of 30 lbs. or more conferred twice the risk of developing gout in comparison with those who maintained weight.32
Fortunately, weight loss appears to mitigate risk. Data from the same cohort show weight loss of greater than 10 lbs. reduces gout risk; these findings taken together provide support for obesity and weight gain as causal factors for gout.32 As more people turn to surgical management for obesity, prospective studies have found a reduction in incident hyperuricemia and UA levels and an increase in recovery from hyperuricemia after bariatric surgery.33,34 Moreover, post-bariatric surgery patients were discovered to have a significant reduction in inflammatory response to monosodium urate crystals with a decrease in peripheral blood mononuclear cell production of IL-1β, IL-8 and IL-6, pointing to the proinflammatory nature of obesity.35
Conclusion
Gout is not a simple inflammatory arthritis, but has complex associations with many aspects of metabolism. The associations between hyperuricemia, gout, the MetS and its components are multi-faceted and may be causal. Further longitudinal evidence may shed light on these relationships. However, interventional studies could help discern whether hyperuricemia and gout in part precipitate metabolic abnormalities leading to development of the MetS and related morbidity. The high prevalence and clinical consequences associated with gout, hyperuricemia and its comorbidities beg for continued investigation into mechanism and treatment.
Lindsey A. MacFarlane, MD, works in the Division of Rheumatology, Immunology and Allergy at Brigham and Women’s Hospital in Boston.
Daniel H. Solomon, MD, MPH, works in the Division of Rheumatology, Immunology and Allergy and the Division of Pharmacoepidemiology and Pharmacoeconomics at Brigham and Women’s Hospital in Boston.
Seoyoung C. Kim, MD, ScD, MSCE, works in the Division of Rheumatology, Immunology and Allergy and the Division of Pharmacoepidemiology and Pharmacoeconomics at Brigham and Women’s Hospital in Boston.
Acknowledgments/Disclosures
- Dr. Kim is supported by the NIH grant K23 AR059677. She received a research grant from Pfizer.
- Dr. Solomon is supported by the NIH grant K24 AR055989. He receives salary grants from research support to Brigham and Women’s Hospital from Amgen, Lilly, Pfizer and CORRONA. He serves in unpaid roles on trials funded by Pfizer, Novartis, Lilly and Bristol-Myers Squibb. He receives royalties from UpToDate.
References
- Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: The National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011;63(10):3136–3141.
- Zhu Y, Pandya BJ, Choi HK. Comorbidities of gout and hyperuricemia in the US general population: NHANES 2007–2008. Am J Med. 2012;125(7):679–687 e1.
- Beltran-Sanchez H, Harhay MO, Harhay MM, McElligott S. Prevalence and trends of metabolic syndrome in the adult U.S. population, 1999–2010. J Am Coll Cardiol. 2013;62(8):697–703.
- Sui X, Church TS, Meriwether RA, et al. Uric acid and the development of metabolic syndrome in women and men. Metabolism. 2008;57(6):845–852.
- Tsouli SG, Liberopoulos EN, Mikhailidis DP, et al. Elevated serum uric acid levels in metabolic syndrome: An active component or an innocent bystander? Metabolism. 2006;55(10):1293–1301.
- Ryu S, Song J, Choi BY, et al. Incidence and risk factors for metabolic syndrome in Korean male workers, ages 30 to 39. Ann Epidemiol. 2007;17(4):245–252.
- Choi HK, Ford ES. Prevalence of the metabolic syndrome in individuals with hyperuricemia. Am J Med. 2007;120(5):442–447.
- Lee JM, Kim HC, Cho HM, et al. Association between serum uric acid level and metabolic syndrome. J Prev Med Public Health. 2012;45(3):181–187.
- Ford ES, Li C, Cook S, Choi HK. Serum concentrations of uric acid and the metabolic syndrome among US children and adolescents. Circulation. 2007;115(19):2526–2532.
- Yang T, Chu CH, Bai CH, et al. Uric acid level as a risk marker for metabolic syndrome: A Chinese cohort study. Atherosclerosis. 2012;220(2):525–531.
- Choi HK, Ford ES. Haemoglobin A1c, fasting glucose, serum C-peptide and insulin resistance in relation to serum uric acid levels—the Third National Health and Nutrition Examination Survey. Rheumatology (Oxford). 2008;47(5):713–717.
- Bedir A, Topbas M, Tanyeri F, et al. Leptin might be a regulator of serum uric acid concentrations in humans. Jpn Heart J. 2003;44(4):527–536.
- Nakagawa T, Tuttle KR, Short RA, et al. Hypothesis: Fructose-induced hyperuricemia as a causal mechanism for the epidemic of the metabolic syndrome. Nat Clin Pract Nephrol. 2005;1(2):80–86.
- Baldwin W, McRae S, Marek G, et al. Hyperuricemia as a mediator of the proinflammatory endocrine imbalance in the adipose tissue in a murine model of the metabolic syndrome. Diabetes. 2011;60(4):1258–1269.
- Rho YH, Zhu Y, Choi HK. The epidemiology of uric acid and fructose. Semin Nephrol. 2011;31(5):410–419.
- Nakagawa T, Hu H, Zharikov S, et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol. 2006;290(3):F625–F631.
- Krishnan E, Akhras KS, Sharma H, et al. Relative and attributable diabetes risk associated with hyperuricemia in US veterans with gout. QJM. 2013;106(8):721–729.
- Dehghan A, van Hoek M, Sijbrands EJ, et al. High serum uric acid as a novel risk factor for type 2 diabetes. Diabetes Care. 2008;31(2):361–362.
- Bhole V, Choi JW, Kim SW, et al. Serum uric acid levels and the risk of type 2 diabetes: A prospective study. Am J Med. 2010;123(10):957–961.
- Krishnan E, Pandya BJ, Chung L, et al. Hyperuricemia in young adults and risk of insulin resistance, prediabetes, and diabetes: A 15-year follow-up study. Am J Epidemiol. 2012;176(2):108–116.
- Nakanishi N, Okamoto M, Yoshida H, et al. Serum uric acid and risk for development of hypertension and impaired fasting glucose or Type II diabetes in Japanese male office workers. Eur J Epidemiol. 2003;18(6):523–530.
- Choi HK, De Vera MA, Krishnan E. Gout and the risk of type 2 diabetes among men with a high cardiovascular risk profile. Rheumatology (Oxford). 2008;47(10):1567–1570.
- Bruderer SG, Bodmer M, Jick SS, Meier CR. Poorly controlled type 2 diabetes mellitus is associated with a decreased risk of incident gout: A population-based case-control study. Ann Rheum Dis. 2014 Apr. 12;[Epub ahead of print].
- Donath MY. Targeting inflammation in the treatment of type 2 diabetes: Time to start. Nat Rev Drug Discov. 2014;13(6):465–476.
- Vitale A, Cantarini L, Rigante D, et al. Anakinra treatment in patients with gout and type 2 diabetes. Clin Rheumatol. 2014 Apr. 15;[Epub ahead of print].
- Choi HK, Zhang Y. Bariatric surgery as urate-lowering therapy in severe obesity. Ann Rheum Dis. 2014;73(5):791–793.
- Juraschek SP, Miller ER 3rd, Gelber AC. Body mass index, obesity, and prevalent gout in the United States in 1988–1994 and 2007–2010. Arthritis Care Res (Hoboken). 2013;65(1):127–132.
- Maynard JW, McAdams DeMarco MA, Baer AN, et al. Incident gout in women and association with obesity in the Atherosclerosis Risk in Communities (ARIC) Study. Am J Med. 2012;125(7):717 e9–717 e17.
- DeMarco MA, Maynard JW, Huizinga MM, et al. Obesity and younger age at gout onset in a community-based cohort. Arthritis Care Res (Hoboken). 2011;63(8):1108–1114.
- Oyama C, Takahashi T, Oyamada M, et al. Serum uric acid as an obesity-related indicator in early adolescence. Tohoku J Exp Med. 2006;209(3):257–262.
- Kim TH, Lee SS, Yoo JH, et al. The relationship between the regional abdominal adipose tissue distribution and the serum uric acid levels in people with type 2 diabetes mellitus. Diabetol Metab Syndr. 2012;4(1):3.
- Choi HK, Atkinson K, Karlson EW, Curhan G. Obesity, weight change, hypertension, diuretic use, and risk of gout in men: The health professionals follow-up study. Arch Intern Med. 2005;165(7):742–748.
- Dalbeth N, Chen P, White M, et al. Impact of bariatric surgery on serum urate targets in people with morbid obesity and diabetes: A prospective longitudinal study. Ann Rheum Dis. 2014;73(5):797–802.
- Sjostrom L, Lindroos AK, Peltonen M, et al. Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. N Engl J Med. 2004;351(26):2683–2693.
- Dalbeth N, Pool B, Yip S, et al. Effect of bariatric surgery on the inflammatory response to monosodium urate crystals: A prospective study. Ann Rheum Dis. 2013;72(9):1583–1584.
