The following day, the patient underwent a bronchoscopy with bronchoalveolar lavage (BAL). Toward the end of the procedure, he developed respiratory distress with significant oxygen desaturation, requiring intubation. The BAL fluid was grossly bloody with a marked elevation of RBCs and WBCs with a neutrophil predominance. There were alveolar macrophages, blood, and acute and chronic inflammatory cells consistent with the diagnosis of diffuse alveolar hemorrhage (DAH).
It’s not unusual for DAH to be the initial manifestation of certain autoimmune and connective tissue diseases, and it’s responsible for many rheumatology consultation requests. DAH is caused by destruction of the alveolar basement membrane or the alveolar capillaries themselves (capillaritis), resulting in accumulation of red blood cells in the alveoli. Clinically, this can lead to hemoptysis, hypoxia, anemia and pulmonary infiltrates on chest X-ray, which can be mistaken for pneumonia.
Pulmonary capillaritis can be isolated to the lung or be a part of a systemic vasculitis, the latter of which can also involve large pulmonary blood vessels. Hemoptysis can be an insidious process or a dramatic presentation causing rapid clinical deterioration. Importantly, hemoptysis can be absent in up to one-third of patients with DAH, and any hemoptysis should be considered potentially life threatening.1
Differential Diagnosis
This patient presented with a multisystem disease process, making the differential diagnosis broad. Based on his elevated ESR and CRP, we know the underlying cause is likely inflammatory or infectious. The biggest clue that vastly narrows the possible causes is hemoptysis in an otherwise healthy patient with no history of lung disease. The major diseases that should be considered include granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), eosinophilic granulomatosis with polyangiitis (eGPA), Goodpasture’s syndrome (GS), systemic lupus erythematosus (SLE) and various infectious etiologies, including tuberculosis. Other less common diseases include isolated pulmonary capillaritis, idiopathic pulmonary hemosiderosis, Behçet’s syndrome, leptospirosis and mixed connective tissue disease.
The Case Continued
A CT scan of the chest showed a right, upper lobe predominant, nodular opacification with central cavitation, with diffuse ground-glass opacities without pleural effusions. Serologic studies revealed negative influenza and blood cultures, and normal C3 and C4 complement levels. Anti-streptolysin and anti-GBM antibodies were negative. Atypical and perinuclear p-ANCA titers <1:20, c-ANCA 1:320, with negative ANA, anti-dsDNA, and anti-Smith antibodies were shown. HIV testing was negative.
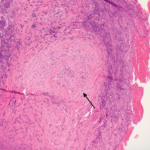
BAL fluid analysis was negative for acid-fast bacilli, viruses, fungi, as well as malignant cells. Following bronchoscopy, the patient was given daily methylprednisolone 1,000 mg for three days, reduced to 80 mg daily for a week, and transitioned to oral prednisone 60 mg per day. Rituximab infusion was started on Day 5 after initiation of immunosuppression, dosed at 600 mg weekly for four weeks. His condition improved dramatically over the next few days, with successful extubation and vastly improved infiltrates on chest X-ray. He developed hoarseness, prompting an ENT evaluation that revealed bilateral vocal process granulomas (contact granulomas). He was discharged on 60 mg prednisone daily with weekly 600 mg rituximab infusions. A presumptive diagnosis of GPA was made, and a definitive diagnosis made after all laboratory results were available some time later.
What Is GPA?
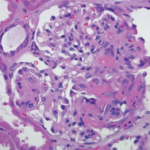
GPA is a rare type of primary vasculitis. The mean age at time of diagnosis is 41 years, and without treatment, mean survival time is less than one year. The presentation and clinical course are variable, ranging from mild disease involving only the upper respiratory tract (i.e., sinuses) to life-threatening involvement causing pulmonary hemorrhage and/or rapidly progressive glomerulonephritis (GN). There is a strong association with c-ANCA and anti-proteinase 3 antibodies.2