Kanda et al. treated a patient who developed repeated relapses of membranous nephropathy related to IgG4-RD initially with prednisolone, but after failure of this approach, treatment cyclophosphamide was added. Due to side effects of cyclophosphamide, the combination of prednisolone and cyclosporine A (CyA) was effective for the induction and maintenance of remission (see Table 7).51
In the event IgG4-RD is refractory or recurrent, the addition of immune-suppressants, such as azathioprine, methotrexate and MMF or rituximab, is recommended. Some patients may relapse after being treated with low doses of azathioprine (50 mg/d) or mycophenolate (1 g/d), but may respond to higher doses of these drugs.52 Rituximab is used to maintain disease remission with glucocorticoid treatment, and successful use of rituximab without associated glucocorticoids has been reported.53 B cell depletion not only interferes with antigen presentation by plasmablasts, it also reduces immune complex formation. The potential significance of immune complex formation as a possible disease mechanism seems relevant for patients with low complement levels— because IgG4 doesn’t bind complement well—and the manifestation of IgG-related TIN.54
In active IgG4-RD, circulating plasmablasts are elevated, so they could be used as a potential biomarker not only for diagnosis, but also for assessment of treatment response. Plasmablasts are circulating plasma cells that arise from activated CD20+ B cells and secrete antibodies. Anti-CD20 monoclonal antibodies function via antibody-dependent cell-mediated cytotoxicity and deplete circulating and tissue resident B cells, thus eliminating the progenitor to the plasmablast. Plasmablasts fall quickly after B cell depletion. The decline or recurrent rise of those cells sometimes correlates better with disease activity than the serum IgG concentration.42
Plasmablast-targeted therapy may offer a more specific treatment approach for IgG4-RD. XmAb5871, a monoclonal antibody with a high-affinity variable region binding to CD19 and an enhanced Fc domain that binds to the FcγRIIb inhibitory receptor of B cells, is currently in phase II development for IgG4-RD treatment and has already been studied in phase I trials in rheumatoid arthritis and systemic lupus erythematosus.55
Oligoclonally-expanded CD4+ effector-memory T cells with a cytotolytic phenotype (CD4+ CTLs) have been characterized recently in IgG4-RD. Those cells express SLAMF7, IL-1β, TGF-β1, granzyme B and perforin, and are the dominant T cells in the affected tissues. After rituximab administration, the concentrations and percentages of these novel T cells with CD4+CTLs phenotype decreased significantly but slower than the concentrations of B cells and plasmablasts. The responsiveness of those novel therapies to CD20-targeted B cell depletion is most likely attributed to the interference of T and B cells, because plasmablasts present antigen and activate memory CD4+CTLs at the inflammation site.56
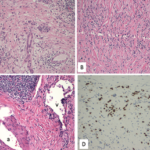
Immunostaining for IgG subclasses wouldn’t distinguish between primary MGN & IgG4-related MGN. However, staining for PLA2R is positive in the GBMs in the case of primary MGN with a sensitivity of 74%.
Conclusions
One of the rare systemic manifestations of IgG4-RD is the kidney. IgG4-RTIN with characteristic imaging findings remains the most common type of kidney involvement in IgG4-RD. Glomerular diseases can develop simultaneously, or as isolated lesions in IgG4-RKD. The most common extra-renal manifestations include AIP, sialadenitis and dacryoadenitis. IgG4 is an important molecule, but its direct role in the disease pathogenesis remains unclear. On the other hand, plasmablasts are potential biomarkers of the disease, because they are found to be elevated in the peripheral blood of patients with IgG4-RD, correlating with disease activity and assisting with the optimal timing of treatment.